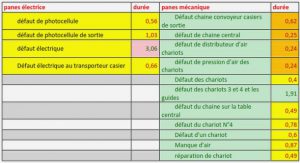
Molécules aimants (Single Molecule Magnet SMM)
Les aimants sont caractérisés par une température critique, température de Curie (T C ). Pour des températures inférieures à T C , ils conservent, après l’application d’un champ magnétique externe, une aimantation, appelée aimantation rémanente. De plus, l’évolution de leur aimantation avec le champ magnétique présente un cycle d’hystérésis (cf. figure 1.2). Un aimant sera d’autant plus intéressant pour des applications que sa température de Curie est élevée. En effet, au-delà de cette température, le matériau retrouve un comportement paramagnétique et l’aimantation rémanente disparaît avec le cycle d’hystérésis.
Ces mêmes caractéristiques, détenues habituellement par des matériaux, sont dans le cas des molécules aimants (Single Molecule Magnet, SMM), reproduites à l’échelle de la molécule. Ellessont en général constituées de plusieurs porteurs de spin (métaux) entourés de ligands organiques.
La première molécule aimant a été présentée en 1993 par R. Sessoli et al. (cf. figure 1.3). Il s’agit d’un assemblage d’ions manganèse aux degrés d’oxydation +II et +III présentant un spin total S=10 dans l’état fondamental et une anisotropie qui résulte du couplage spin-orbite et provoque une levée de dégénérescence partielle de l’état fondamental en l’absence de champ extérieur. Lorsqu’un champ externe est appliqué, tous les spins s’alignent et seul le niveau caractérisé par une projection de spin Ms = 10 est peuplé. C’est l’équivalent de la première aimantation des matériaux. L’anisotropie empêche alors la relaxation du système qui garde son aimantation une fois le champ coupé (cf. figure 1.3(b) et 1.4), pour une température inférieure à 4 K.
Les SMM sont l’objet de nombreuses études expérimentales et théoriques.
L’enjeu essentiel est de concevoir des molécules ayant un état fondamental haut-spin et une grande barrière anisotropique empêchant la relaxation de la magnétisation induite en première aimantation. L’anisotropie, i.e. l’aptitude du système à se comporter différemment selon la direction du champ magnétique appliqué, résulte de la distorsion de la sphère de coordination du complexe par rapport à une géométrie cubique et de l’effet du couplage spin-orbite.
Récemment, l’étude théorique des effets du couplage spin-orbite sur la spectroscopie a en partie mis à jour les mécanismes qui régissent l’anisotropie d’un édifice complexe.
Les éléments impliqués ici sont nul, les niveaux sont également peuplés. (b) A la première aimantation, un niveau se peuple plus que l’autre. (c) Lors du retour au champ nul, la barrière anisotropique bloque la relaxation du système. Cette dernière évolue proportionnellement au produit de D, un paramètre qui quantifie l’anisotropie d’un système, par S2. complexes et ne constituent pas l’objectif de ce travail. Mais ils représentent un exemple où les outils de la chimie quantique ont ouvert la voie vers un modèle et une interprétation des propriétés expérimentales de ces systèmes.
Transitions et coopérativité
La première transition thermique abrupte décrite en 1964 (cf. figure 1.6), préfigure l’émergence de différents types de transition possible. Ainsi, en 1976, E. König et G. Ritter présentent le premier composé dont le relevé du moment magnétique en fonction de la température présente un cycle d’hystérésis (cf. figure 1.9(a)). Depuis, ce phénomène s’est retrouvé dans de nombreux composés pouvant présenter des largeurs de cycle allant jusqu’à 90 K.
Un dernier type de transition fut révélé en 1982 avec l’étude de [Fe(2-pic) 3 ]Cl 2 .EtOH (pic=picolylamine) qui présente une transition de spin en deux étapes (cf. figure 1.9(b)). Dix ans plus tard, ce même palier fut exhibé pour un composé dinucléaire.
Modélisations thermodynamiques de la transition de spin
La coopérativité est une manifestation indéniable du caractère collectif de la transition de spin. Seul le traitement d’un assemblage de molécules va pouvoir identifier et quantifier les interactions entre sites responsables des propriétés macroscopiques expérimentales observées. Ainsi, des modèles thermodynamiques ont été élaborés. Deux sont présentés ici, le modèle de Wajnflaszqui consiste en une approche moléculaire de la transition de spin, puis le modèle des solutions régulières proposé par C. P. Slichter et H. G. Drickamer.
Apport de la chimie quantique à la transition de spin
L’approche champ de ligands a permis, dans la section 1.2.2, d’expliquer de façon très efficace le phénomène de transition de spin. En effet, avec ce modèle, les paramètres qui gouvernent la transition sont mis en lumière, les conséquences de la transition sur la structure ou la spectroscopie du système également. Un autre avantage de cette approche paramétrée est de livrer une image intelligible de la transition quand les méthodes de chimie théorique, basées sur la densité électronique ou sur la fonction d’onde, conservent un caractère mystérieux. Toutefois, la modélisation thermodynamique des systèmes à transition de spin repose essentiellement, ainsi que le présente la section 1.2.4, sur des paramètres moléculaires (cf. figure 1.10) que seules les méthodes de chimie quantique peuvent atteindre.
Un premier défi à relever est l’élucidation de la géométrie moléculaire des complexes à transition de spin qui échappe parfois aux inspections expérimentales (résumé sur la figure 1.10 par les distances dbs et dhs). Viennent ensuite les paramètres énergétiques (positionnement respectif des états BS et HS). L’inspection des vibrations moléculaires représente un enjeu primordial puisqu’elles donnent accès à la partie essentielle de l’entropie. Enfin vient l’étude des autres propriétés spectroscopiques (vibrations, paramètre Mössbauer, Raman, etc.). Ces données calculéesà l’échelle de la molécule ont fait l’objet de nombreuses études qui seront présentées dans le chapitre 3.
Bistabilité rédox
La première évocation de l’utilisation de composés possédant une activité rédox dans le domaine de l’électronique moléculaire par A. Aviram et M. A. Ratner est apparue en 1974.
L’idée d’introduire au sein d’un domaine consacré entièrement à la circulation d’électrons, des systèmes qui présentent une sensibilité à ces mêmes électrons paraît, en effet, assez naturelle. Malgré cette affinité originelle, il faudra attendre la fin des années 1990 avant qu’émergent des systèmes mettant à profit l’activité rédox pour l’électronique moléculaire.
Dans cette partie, quelques fonctions remplies par des molécules grâce à leur activité rédox sont présentées par le biais d’exemples. Ensuite, l’importance des porphyrines au sein de cette famille est soulignée.
Commutateurs moléculaires
Les commutateurs remplissent une fonction essentielle puisqu’ils permettent de former les portes logiques nécessaires dans les efforts de programmation. La réduction au niveau molécu-33 laire de cette fonction est donc un passage obligé vers la nanoélectronique. Ils sont également utiles dans les composants mémoires de type « crossbar ».
La réduction à l’échelle moléculaire repose sur un montage métal – molécule – métal qui doit posséder (au moins) deux états stables, l’un présentant une résistance importante, l’autre une résistance faible.
En 1999, C. P. Collier et al. présentent un tel agencement basé sur l’utilisation des rotaxanes (cf. figure 1.11). Les rotaxanes consistent en deux, ou plus, parties imbriquées entre elles. Une de ces parties présente une courbure, des sites fonctionnalisés, ainsi que deux butées à ses extrémités. Le second élément est constitué d’un anneau qui se retrouve piégé autour de la première partie. Dans la mesure où aucun lien covalent ne vient se placer entre les deux parties du rotaxane, l’anneau est libre de se déplacer sous l’influence d’un paramètre, ici le degré d’oxydation de la molécule.
Approche quantique de la structure électronique
Compte-tenu de leur implication dans de nombreux mécanismes biologiques ainsi que le nombre important d’applications possibles, les porphyrines ont été le centre d’une intense activité de calcul.
Toutefois, les propriétés intrinsèques des porphyrines, qui font leur intérêt, causent également de nombreuses difficultés dans leur modélisation. Ainsi, les optimisations de géométrie de ces composés sont souvent délicates, une conséquence de la grande flexibilité de ces ligands.
La labilité électronique de ces composés ainsi que la présence de couches ouvertes rend les approches DFT parfois inopérantes.
Les stratégies employées pour surmonter ces difficultés seront présentées dans les chapitres 2 et 4.
Tautomérisme de valence
Les contributions à la bistabilité des systèmes à transition de spin d’une part et des systèmes rédox d’autre part s’est prolongée avec l’émergence il y a trente ans de composés réunissant le transfert de charge et le changement de spin, les complexes à tautomérisme de valence.
Définitions
Les complexes à tautomérisme de valence combinent des ligands possédant une activité rédox et des métaux de transition avec au moins deux degrés d’oxydation accessibles. Ils possèdent deux états électroniques proches en énergie et une structure électronique localisée. L’environnement conditionne grandement la distribution de charge dans le système et, à l’image du phénomène de transition de spin, une perturbation extérieure peut accomplir une conversion réversible entre les deux états électroniques. Le premier exemple de complexe à tautomérisme de valence a été présenté en 1980 par R. M. Buchanan et C. G. Pierpont. Il s’agissait d’un complexe de cobalt associé avec un ligand quinone.
Cette chimie reste à la base de la plupart de ces complexes, même si de nouvelles voies sont apparues après les années 2000.
Un système modèle
Nous avons étudié cette dépendance sur un complexe modèle [Fe(NCH)6 ] 2+ pris dans une géométrie octaédrique (cf. figure 2.11). Les atomes Fe, N, C et H sont décrits par des bases de type ANO-RCC avec les contractions respectives [7s6p5d3f 2g1h], [4s3p1d], [3s2p1d] et [1s]. L’espace actif choisi contient les 5 orbitales d du fer, un second jeu d’orbitales d comptant un nœud supplémentaire et 2 orbitales de type σ entre le fer et ses ligands. Au final, l’espace actif compte 10 électrons pour 12 orbitales (CAS[10,12]). Les choix des bases et de l’espace actif sont détaillés dans le chapitre suivant.
Deux géométries d’équilibres sont décrites pour [Fe(NCH)6 ] 2+ : l’une correspond à un état bas spin (BS) et des distances Fe–N de 1,90 Å, l’autre à un état haut spin (HS) pour des distances Fe–N de 2,15 Å. Les énergies des états BS et HS, chacun pris dans sa géométrie la plus favorable, ont été calculées au niveau CASPT2 pour différentes valeurs de ǫ. Comme attendu, l’état HS qui présente un caractère couche ouverte plus important est bien plus sensible au paramètre IPEAque l’état BS (cf. figure 2.12).
Ne pas jeter le bébé avec l’eau du bain
Ces études DFT et CASPT2 ont un caractère décourageant quant à la fiabilité des calculs dans l’approche des propriétés de complexes de métaux de transition. Toutefois, il faut rappeler que le cas choisi ici représente l’un des pires scénario envisageables avec un changement de spin S = 0 → S = 2 associé à une modification de la géométrie. En particulier, l’influence de l’IPEA est probablement moins importante sur des phénomènes impliquant un changement plus doux du nombre de couches ouvertes. De plus, comme l’indique la figure 2.14 le comportement énergétique est reproductible d’un complexe à l’autre. Ainsi sans avoir la valeur exacte du gap adiabatique, les tendances peuvent tout de même être étudiées avec confiance.
Le message qui doit rester est une mise en garde contre l’utilisation « boîte noire » qui pourrait être faite des méthodes de chimie quantique dites ab initio qui ne le sont pas tout à fait.
Apport des orbitales locales
Description
Bien avant l’émergence des orbitales moléculaires, le problème de la liaison chimique s’était posé et avait trouvé une réponse dans la description proposée par G. N. Lewis.
Cette dernière consiste en une description de la valence, les liaisons étant décrites comme la mise en commun des électrons de valence de chacun des atomes. Ainsi les liaisons possèdent un fort caractère local. Les méthodes de calcul Valence Bond sont directement issues de ces approches locales de la structure électronique des molécules. Elles souffrent malheureusement de la manipulation d’orbitales non-orthogonales. C’est pourquoi les orbitales moléculaires, qui autorisent une délocalisation arbitraire mais permettent de former un jeu orthogonal plus simple à manipuler, se sont imposées dans la description quantique des liaisons chimiques. Ainsi l’efficacité du calcul l’a emporté sur la puissance d’analyse que possède la structure de Lewis. Au début des années 2000, D. Maynau et al. réintroduisent le concept d’orbitales locales avec l’objectif de tirer profit du caractère local de la corrélation électronique.
En effet, les effets de la corrélation sont d’autant plus importants qu’ils impliquent des orbitales proches l’une de l’autre dans l’espace. Ainsi, les déterminants excités proposant le transfert d’un électron d’une partie de la molécule vers une autre éloignée de la première auront un poids faible dans la fonction et pourront être négligés sans affecter le résultat. On peut alors réduire l’espace de configurations pour incorporer la corrélation dynamique. En plus de leur force interprétative, les orbitales locales permettent d’affronter des systèmes de taille plus importante en limitant la considération des déterminants excités porteurs d’un réel sens physique.
|
Table des matières
Introduction générale
1 Systèmes moléculaires bistables et hystérétiques
Introduction
1.1 Molécules aimants (Single Molecule Magnet SMM)
1.2 Complexes à transition de spin
1.2.1 Un peu d’histoire
1.2.2 Description du phénomène
1.2.3 Transitions et coopérativité
1.2.4 Modélisations thermodynamiques de la transition de spin
1.2.5 Apport de la chimie quantique à la transition de spin
1.3 Bistabilité rédox
1.3.1 Commutateurs moléculaires
1.3.2 Hystérèse induite par la molécule
1.3.3 Hystérèse moléculaire
1.3.4 Les porphyrines
1.4 Tautomérisme de valence
1.4.1 Définitions
1.4.2 Manifestations du tautomérisme de valence
Conclusion
Bibliographie
2 Aspects méthodologiques
Introduction
2.1 Traitement du problème polyélectronique
2.1.1 Le problème polyélectronique
2.1.2 Construction de la fonction d’onde
2.1.3 Une première réponse : la méthode Hartree-Fock
2.1.4 Limites de la méthode Hartree-Fock : H 2
2.2 La corrélation électronique
2.2.1 Corrélation statique et corrélation dynamique
2.2.2 Une approche de la corrélation statique : le méthode CASSCF
2.2.3 La corrélation électronique à l’œuvre : l’exemple de (H 2 )2
2.2.4 Traitements post-CASSCF de la corrélation dynamique
2.2.5 Les approches DFT
2.2.6 Des méthodes ab initio ?
2.3 Apport des orbitales locales
2.3.1 Description
2.3.2 Force interprétative des orbitales locales
Conclusion
Bibliographie
Publications liées
3 Phénomène d’hystérèse dans les composés à transition de spin
Introduction
3.1 Approches de la coopérativité
3.2 Inspections microscopiques
3.2.1 Performances des approches DFT et CASPT2
3.2.2 Etude ab initio d’un complexe modèle : [Fe(NCH) 6 ] 2+
3.2.3 Redistribution électronique dans les complexes à transition de spin
3.2.4 Champ du cristal versus champ de ligands
3.3 Un modèle électrostatique
3.4 Importance des effets de polarisation
3.4.1 Confrontation avec des systèmes synthétiques
3.4.2 Comment maîtriser Γpol?
3.4.3 Histoire d’une réconciliation
Conclusion
Bibliographie
Publications liées
4 Inspections ab initio de complexes de porphyrine
Introduction
4.1 Description quantique des porphyrines de manganèse(II)
4.1.1 Position du problème
4.1.2 Détails des calculs
4.1.3 Performances comparées des méthodes CASPT2 et DFT
4.2 Cas des porphyrines de manganèse(III)
4.2.1 Spectroscopie de basse énergie de [MnPCl]
4.2.2 Oxydation des complexes de manganèse(II) : Mn(III)P + vs. Mn(II)P•+
4.3 Scénario pour une hystérèse moléculaire
Conclusion
Bibliographie
Publication liée
5 Approche semi-locale de systèmes délocalisés
Introduction
5.1 L’approche semi-locale
5.1.1 Construction de la fonction d’onde N -électronique
5.1.2 Hamiltoniens modèles
5.1.3 Energie de cohésion
5.1.4 SCPE et EPV
5.2 Energie de cohésion de systèmes périodiques
5.2.1 Systèmes régis par l’hamiltonien de Hückel
5.2.2 Systèmes régis par l’hamiltonien de Hubbard
5.3 Peierls et Little
5.3.1 Etude de la dimérisation en Hubbard
5.3.2 Suppression de la distorsion suivant la conjecture de Little
5.4 Ouverture du gap de charges
5.4.1 Etude d’un dimère corrélé
5.4.2 Energie de cohésion
5.4.3 Approche excitonique du gap de charge
5.4.4 Résultats préliminaires
Conclusion
Bibliographie
Publication liée
Conclusion générale