Télécharger le fichier pdf d’un mémoire de fin d’études
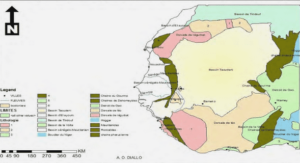
Répartition géographique de la drépanocytose
La drépanocytose est une maladie extrêmement répandue, surtout en Afrique Noire Sub-saharienne.
La fréquence du trait drépanocytaire augmente d’Ouest en Est et du Nord au Sud de l’Afrique. On appelle ‘’ceinture sicklémique’’ la zone qui s’étend entre le 15ème parallèle, latitude Nord et le 20ème parallèle, latitude Sud, et c’est dans cette zone que les populations noires sont les plus atteintes. On passe de 5-20% de porteurs du trait drépanocytaire en Afrique de l’Ouest, à 40% et plus chez certaines ethnies de l’Afrique Centrale (République Démocratique du Congo, Congo, Gabon). La drépanocytose est également très répandue chez les Noirs américains (9% aux USA), chez les antillais (12%), aux Caraïbes, en Amérique du Sud, (notamment au Brésil) (Mbeyeme et col, 2004) (Figure 4).
D’après Fleming, environ 70 millions de personnes dans le monde possèdent le trait drépanocytaire, parmi lesquelles 58 millions vivent en Afrique, surtout entre le sud du Sahara et le nord de la rivière Zambèze. Près de 150 000 enfants naissent chaque année dans le monde avec la drépanocytose, environ 120 000 de ces enfants sont Africains. On retrouve également le trait drépanocytaire, mais plus rarement, chez certaines populations non-noires du Bassin Méditerranéen (Maghreb, Italie, Turquie), du Moyen Orient, de l’Inde (Fleming, 1989).
La répartition géographique du trait drépanocytaire est grossièrement superposable aux zones d’endémie à Plasmodium falciparum. Il semble que le plasmodium se développe moins bien dans les hématies HbSS, d’où une sélection naturelle qui s’est produite pendant des millénaires. L’anomalie génétique est un avantage sélectif qui pourrait expliquer sa persistance dans les régions d’endémie palustre, alors que les formes homozygotes sont le plus souvent mortelles. La présence d’hémoglobine S n’empêche pas la multiplication du plasmodium, mais limite notablement la fréquence des formes graves avec neuropaludisme (Sanokho et col., 1984).
Au Sénégal, la prévalence du trait drépanocytaire est estimée entre 8% et 10% (Mbodji et col, 2003).
Une étude chez les nouveaux nés qui sont du reste très exposés à l’affection du paludisme, a permis la découverte de 0,4% de formes homozygotes à Dakar (Diop et col., 2010).
Mode de transmission génétique
Si deux personnes hétérozygotes AS veulent avoir des enfants, ils courent à 25% le risque à chaque grossesse de donner naissance à un enfant drépanocytaire (SS) ou à un enfant sain (AA) au même pourcentage et à 50% il risque de donner naissance à un enfant porteur du trait drépanocytaire (Labie et col, 2003) (figure 5).
Physiopathologie de la drépanocytose
Rappel sur l’hémoglobine
L’hémoglobine normale est une chromoprotéine porphyrique de coloration rouge renfermant du fer. Contenue dans le globule rouge circulant, l’Hb est le transporteur de l’oxygène de l’air vers les tissus ce qui permet une bonne oxygénation de ces derniers. Chaque molécule d’hémoglobine est formée de quatre sous-unités identiques deux à deux et chaque sous-unité est formée d’un noyau d’hème et d’une globine. L’ensemble de la structure est stabilisée grâce à des liaisons de faible énergie établies entre les différentes structures (Russel, 1996) (figure 6).
Plusieurs hémoglobines se succèdent au cours de la vie et à tout moment, il en existe plusieurs simultanément. Ces hémoglobines se distinguent par la nature des sous-unités qui les constituent (figure 7). Chez l’être humain, au cours de l’évolution ontogénique, le profil des hémoglobines change deux fois.
La première de ces commutations (ou « switch ») coïncide avec le passage de la vie embryonnaire à la vie fœtale, la seconde avec celui de la vie fœtale à la vie adulte (Wajcman, 2005) (figure 7). Ces commutations sont à l’origine de la différence des hémoglobines embryonnaires, fœtales et adultes (Figure 8). Durant la vie embryonnaire, on distingue l’Hb Gower 1 (δ2ε2), l’Hb Gower 2 (α2ε2) et l’Hb Portland (δ2γ2). L’hémoglobine fœtale (Hb F) de structure α2γ2 est détectable à partir de la 5ème semaine de vie intra-utérine. Chez l’adulte, plus de 95% de l’hémoglobine est de type A1 (α2β2). L’hémoglobine A2 (α2δ2) ne dépasse pas 3% chez un sujet sain (Wajcman, 2005).
Physiopathologie proprement dite
Trois principaux phénomènes sont impliqués dans la physiopathologie de la drépanocytose :
Polymérisation de l’hémoglobine S et falciformation du globule rouge
Déshydratation du globule rouge
Interaction des globules rouges falciformes avec l’endothélium vasculaire
Polymérisation de l’hémoglobine S et falciformation du globule rouge
La polymérisation de l’hémoglobine s’explique par le fait que la valine 6 qui est un résidu hydrophobe remplace un acide aminé hydrophile, l’acide glutamique. Les globines étant entourées par un film d’eau, la présence d’un site hydrophobe crée un point de « collage » entre 2 molécules d’hémoglobine voisines. Celui-ci s’établit entre la leucine 88 et la phénylalanine 85 d’une chaîne alpha d’une molécule d’hémoglobine et la valine 6 de la chaîne β de l’hémoglobine voisine, d’où la création d’une structure cristalline en fibres. Les dimères HbS/HbS s’assemblent pour former des brins ; les brins s’associent en fibres, responsables de la déformation des hématies. La polymérisation est un processus coopératif qui demande un certain délai d’initiation. Il y a donc une course de vitesse entre le temps de passage de l’hématie dans ce goulot d’étranglement qu’est le capillaire et le délai de polymérisation qui transforme un globule flexible en une particule rigide et donc susceptible de rester bloquer.
La succession dans le temps des cycles de polymérisation dépolymérisation aboutit à la modification des caractéristiques physiques du globule rouge lui conférant un aspect en faucille : c’est le phénomène de falciformation caractéristique de la drépanocytose. Elle s’accompagne de modifications majeures de la membrane cellulaire du globule rouge.
La falciformation est au début réversible et disparaît lors de la réoxygénation des globules rouges. Après plusieurs cycles de polymérisation, les lésions membranaires conduisent de façon irréversible à la formation des globules rouges falciformes. Les globules rouges, déformés par ces structures fibreuses tubulaires prennent une forme caractéristique en faucille ou feuille de houx, appelés drépanocytes (Segbena et col., 1994) (figure 9).
Déshydratation du globule rouge
La falciformation du globule rouge est réversible en cas de réoxygénation. Mais au bout d’un certain temps, les lésions de la membrane érythrocytaire sont à l’origine de la déshydratation, puis de la falciformation irréversible de ce globule rouge. Chez le drépanocytaire, on observe une sortie anormalement élevée de H2O et d’ions K+ (Gardos, 1958). Deux canaux sont impliqués dans ces phénomènes de déshydratation (figure 10) : Le cotransporteur K-Cl et le canal Gardos (canal K+ activé par le Ca2+).
Les drépanocytes ont une durée de vie de 10 à 15 jours tandis que les globules rouges normaux ont une durée de vie de 120 jours.
Cela s’explique par le fait que les drépanocytes sont fragiles et sont plus rapidement détruits au niveau de la microcirculation splénique (Lefrère, 2005).
Interaction des globules rouges falciformes avec l’endothélium vasculaire
Il a été montré qu’il y avait un processus d’adhérence anormale des hématies falciformées aux cellules de l’endothélium vasculaire suggérant des altérations rhéologiques chez les patients atteints de la drépanocytose (Hoover et col., 1979).
De plus, l’enchaînement des cycles de falciformation/défalciformation des globules rouges modifient leur potentiel adhésif en augmentant l’expression de certains récepteurs des molécules d’adhésion (Chiang et col, 2005). Par ailleurs, les sujets drépanocytaires ont un environnement vasculaire pro-inflammatoire propice à l’adhérence des hématies falciformes et des leucocytes. Les leucocytes des patients atteints de la drépanocytose ont également un potentiel adhésif plus important que ceux des sujets sains (Kaul et col, 2006). L’adhésion à l’endothélium se fait via l’expression des protéines pro-adhésives spécifiques telles que VCAM-1 (Vascular Cell Adhesion Molecule-1), ICAM-1 (Intercellular Cell Adhesion Molecule-1), P-sélectine et E-sélectine (Manwani , 2013). Il y a donc un enchaînement d’évènements chez les drépanocytaires qui conduit aux crises vaso-occlusives :
les phénomènes d’adhésion vasculaire ralentissent le flux sanguin et permettent la falciformation des érythrocytes ;
les hématies falciformées obstruent les microvaisseaux et ces obstructions vasculaires créent une hypoxémie locale, ce qui favorise la polymérisation de l’Hb S.
Le facteur déclenchant une crise vaso-occlusive est donc souvent difficile à identifier. Cependant, lors des crises vaso-occlusives, on observe fréquemment chez les drépanocytaires une surexpression des molécules d’adhésion (VCAM-1, ICAM-1, P-sélectine et E-sélectine) par les cellules endothéliales (Solovey et col., 1997). Cette adhésion importante proviendrait donc de multiples activations cellulaires (Kaul et col, 1993) (figure 11).
Symptomatologie
La symptomatologie décrit l’ensemble des signes de la maladie. Ainsi nous distinguons les signes cliniques et les signes biologiques.
Signes cliniques
Les signes cliniques sont polymorphes et varient en fonction de la forme génétique de la maladie.
Chez le sujet drépanocytaire de forme SS
Les signes cliniques de la maladie drépanocytaire apparaissent après le premier trimestre de la vie, lorsque l’hémoglobine fœtale F est progressivement remplacée par l’hémoglobine S. Ces signes sont variables d’un patient à un autre, selon l’évolution de la maladie on distingue deux phases : la phase intercritique et la phase critique
Phase intercritique
Elle est caractérisée par la triade symptomatique MINKOWSKI-CHAUFFARD qui associe l’anémie, l’ictère et la splénomégalie. La pâleur cutanéo muqueuse, expression de l’anémie hémolytique chronique, est une manifestation constante et précoce de la drépanocytose.
L’ictère cutanéo muqueux de type hémolytique est caractérisé par des urines foncées non mousseuses et des selles de couleur normale. La splénomégalie est fréquente chez le nourrisson et le jeune enfant. Son volume va diminuer progressivement au cours des années pour disparaître dans la plupart du temps au-delà de l’âge de 5 ans. L’aspect général de l’enfant mal suivi est dysmorphique. Le corps est longiligne, les extrémités sont effilées avec une tête volumineuse auxquels sont associés volontiers un retard staturo-pondéral et pubertaire (Sherman, 1940).
Phase critique
Cette phase se caractérise par la survenue de crises vaso-occlusives. C’est la manifestation la plus fréquente et essentiellement la plus douloureuse. Les premières douleurs débutent autour de 6 mois dans l’enfance.
La douleur traitée ou non, est présente pendant une heure à 6 jours avec une durée moyenne de 3 à 4 jours. La période de relative accalmie sera interrompue au bout de quelques temps à périodicité variable, par une nouvelle crise.
Cette répétition de la douleur est caractéristique de la drépanocytose et le siège de cette douleur est également variable selon l’âge du patient.
Chez le nourrisson, la douleur réalise le syndrome ‘’pied main’’ (hand foot syndrome). Elle se manifeste par une nécrose ischémique des petits os des mains et des pieds accompagnée d’une tuméfaction aigue des parties molles du dos des mains et des pieds, dans un contexte fébrile ;
Chez les enfants et les adolescents, la douleur est abdominale ou ostéo-articulaire. Ces douleurs sont provoquées par des infarctus viscéraux (mésentère, rate, foie) et sont souvent accompagnées de fièvre. Elles peuvent simuler un tableau d’abdomen chirurgical sévère ;
Chez l’adulte, la douleur est ostéo-articulaire et siège au niveau des os longs, de ceux du thorax et du bassin (Mitchell BL, 2007).
Chez le sujet drépanocytaire de forme AS
Les sujets ayant une drépanocytose hétérozygote sont aussi appelés porteurs sains de la drépanocytose, ou porteurs du trait drépanocytaire. Ils ont reçu le gène normal A de la β-globine d’un de leurs parents, et le gène drépanocytaire S de l’autre. Le taux d’Hb S est inférieur à 50% (Deme, 2007).
Signes biologiques
Chez l’homozygote (forme SS)
Le test d’Emmel montre la présence de plusieurs drépanocytes.
La drépanocytose homozygote est caractérisée par un taux d’hémoglobine situé entre 7 et 9 g/dl, une réticulocytose entre 200 000 et 600 000/mm3, un volume globulaire moyen normal (VGM). Elle peut aussi être caractérisée par la présence constante sur le frottis sanguin de drépanocytes, une hyperleucocytose à polynucléaires neutrophiles pouvant atteindre 30 000/mm3 sans infection et une tendance à la thrombocytose. L’électrophorèse de l’hémoglobine met en évidence la présence d’hémoglobines S, A2 et F (non systématique) ; il n’y a pas d’hémoglobine A.
Chez l’hétérozygote (forme AS)
Les caractéristiques hématimétriques du sang périphérique des patients drépanocytaires hétérozygotes sont identiques à celle du sang normal. Tant en ce qui concerne la lignée érythrocytaire que les lignées leucocytaires et plaquettaires. La morphologie des hématies est normale et il n’y a pas de drépanocytes en circulation au frottis sauf en cas de crises. Cependant, lorsque les hématies sont incubées dans un milieu privé d’oxygène (test d’Emmel), le phénomène de falciformation se manifeste et fait apparaître des drépanocytes.
Une microcytose constatée chez un drépanocytaire hétérozygote doit faire penser à une carence martiale ; une macrocytose doit suggérer une carence vitaminique, notamment en acide folique ou en vitamine B12.
L’électrophorèse de l’hémoglobine montre une fraction majeure d’hémoglobine A de 55 à 60%, une fraction importante d’hémoglobine S de 40 à 45% et enfin un constituant mineur d’hémoglobine A2 de 2 à 3%. Cet état doit être différencié du sujet drépanocytaire homozygote transfusé ; dans cette situation, l’hémoglobine A du donneur disparaît de l’électrophorégramme dans les semaines qui suivent la transfusion (Hazoumé, 1984).
Complications de la drépanocytose
Les complications sont liées aux accidents vaso-occlusifs et à l’anémie. Elles sont dominées par des thromboses vasculaires et des infections. Ces complications peuvent être aiguës ou chroniques.
Complications aiguës
Accidents vaso-occlusives graves
Accidents vasculaires cérébraux
Ce sont des accidents très graves, souvent mortels qui laissent des séquelles considérables. En effet, il s’agit de thromboses au niveau des artères cérébrales, parfois récidivantes et bilatérales qui se traduisent cliniquement par des céphalées et des crises convulsives. Ces dernières sont accompagnées parfois de troubles du comportement qui peuvent entrainer une hémiplégie, et éventuellement une aphasie, c’est à dire un syndrome pyramidal (Habibi, 2004).
Le syndrome thoracique aigu
Le syndrome thoracique aigu est une pathologie potentiellement grave, caractérisée par la survenue d’une douleur thoracique associée à une symptomatologie pulmonaire à type de dyspnées, accompagnée d’expectoration et de fièvre. A ce stade de la maladie il s’agit d’un foyer pulmonaire clinique ou radiologique, associé dans 50 % des cas à un épanchement pleural. L’insuffisance respiratoire chronique et l’hypertension artérielle pulmonaire peuvent se développer à la suite d’épisodes de syndromes thoraciques (Parot, 2003).
Le Priapisme
Il désigne un état d’érection prolongé. Il s’agit d’une complication fréquente de la drépanocytose homozygote. Le priapisme est une complication grave, sur le plan fonctionnel, de la drépanocytose qui touche 6% des enfants et 42 % des adultes (Habibi, 2004).
Les complications infectieuses
Les complications infectieuses constituent la principale cause de morbidité et de mortalité des drépanocytaires homozygotes d’une manière générale, et en particulier chez l’enfant entre 6 mois et 5 ans. Cette pathologie infantile a tendance à régresser à l’âge adulte.
Les infections qui résultent de ces complications peuvent être :
-bactériennes : c’est-à-dire responsable de méningites et de septicémie, d’ostéomyélite aigue, d’infections pulmonaires, digestives, oculaires et cutanées.
– parasitaires : c’est-à-dire essentiellement représentées par le paludisme d’une gravité particulière.
-virales : dans ce cas, l’infection à parvovirus B19 occasionne chez le drépanocytaire une anémie par érythroblastopénie aigue lors de la virémie Tarer V, 2006).
|
Table des matières
INTRODUCTION
PREMIERE PARTIE : REVUE BIBLIOGRAPHIQUE
Chapitre I : Rappels sur la Drépanocytose
I-Généralités
I-1.Historique
I-2. Répartition géographique de la drépanocytose
I-3. Mode de transmission génétique
II-Physiopathologie de la drépanocytose
II-1. Rappel sur l’hémoglobine
II-2. Physiopathologie proprement dite
II-2.1.Polymérisation de l’hémoglobine S et falciformation du globule rouge
II-2.2. Déshydratation du globule rouge
II-2.3. Interaction des globules rouges falciformes avec l’endothélium vasculaire
III- Symptomatologie
III-1. Signes cliniques
III-1.1. Chez le sujet drépanocytaire de forme SS
III-1.1.1. Phase intercritique
III-1.1.2. Phase critique
III-1.2. Chez le sujet drépanocytaire de forme AS
III-2. Signes biologiques
III-2.1. Chez l’homozygote ( forme SS)
III-2.2. Chez l’hétérozygote (forme AS)
IV- Complications de la drépanocytose
IV-1. Complications aiguës
IV-1.1. Accidents vaso-occlusives graves
IV-2. Complications chroniques
IV-2.1. Rétinopathie
IV-2.2. Ulcère de jambe
V- Diagnostic biologique de la drépanocytose
V-1. Tests de dépistage
V-1-1. Test d’Emmel
V-1.2. Test d’Itano ou test de solubilité
V-2. Test de confirmation
V-2.1. Electrophorèse de l’hémoglobine
V-2.1.1. Electrophorèse sur acétate de cellulose à pH alcalin
V-2.1.2. Electrophorèse sur agar à pH acide
V-2.2. Isoélectrofocalisation
V-2.3.Chromatographie liquide à haute performance (CLHP)
VI- Traitements de la drépanocytose
VI.1. Médecine moderne
IV.1.1. Antalgiques
IV.1.2. Traitements adjuvants
IV.1.3. Transfusion ou échange transfusionnel
IV.1.4. Antifalcémiants
IV.1.5. Hydroxyurée
IV.1.6. Allogreffe de moelle osseuse
IV.1.7. Traitement préventif des infections
IV.1.8. Mesures hygiéno-diététiques
IV.2. Phytothérapie
Chapitre II : Revue bibliographique Hibiscus sabdariffa
I. Généralités sur Hibiscus sabdariffa
I.1. Dénominations
I.2. Classification
I.3. Répartition géographique et Habitat
I.4. Description de la plante
I.5. Composition chimique
I.5.1. Calices
I.5.2. Graines
I.5.3. Feuilles
I.6.Propriétés pharmacologiques
I.6.1. Activité antioxydante
I.6.2. Activité antihypertensive et cardioprotectrice
I.7. Utilisations
I.7.1.Utilisations médicinales
DEUXIEME PARTIE : TRAVAIL EXPERIMENTAL
I-Objectif du travail
II- Cadre de l’étude
III- Matériel et méthodes
III-1. Matériel
III-1.1. Matériel végétal
III-1.2.Matériel de laboratoire
III.1.3. Solvants et Réactifs
III.2. Méthodes
III.2.1. Extraction des composés phénoliques
III.2.3. Extraction des saponosides
IV. Dosage des composés phénoliques totaux
V. Détermination de l’activité antifalcémiante des extraits de H. sabdariffa
V.1. Préparation des solutions
V.1.1. Préparation de la solution tampon phosphate pH 7,4
V.1.2. Préparation des extraits à tester
V.2. Collecte de sang
V.3. Recherche de l’activité antifalciformante
V .3.1. Protocole du test
VI. Résultats et Discussion
VI.1. Résultats
VI.1.1. Rendement des extractions
VI.1.2. Résultats du dosage des composés phénoliques totaux
VI.1.3. Résultats des tests biologiques
IV.2. Discussion
CONCLUSION
REFERENCES BIBLIOGRAPHIQUES
Télécharger le rapport complet