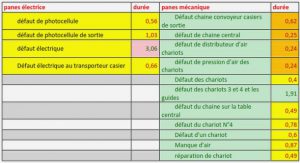
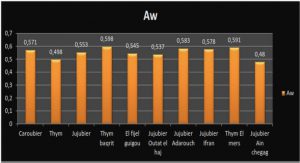
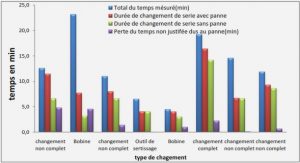
Télécharger le fichier pdf d’un mémoire de fin d’études
Effet de l’ajout d’ions modificateurs sur l e réseau silicaté
Les verres à base de silice et de fondant (CaO, Na 2O…), bien que simples en terme de composition, sont les plus étudiés. Dans notre quotidien par exemple, les verres de composition de base sodo-calciques silicatés sont les plus utilisés (pour les vitres par exemple) du fait de leur faible coût et des températures d’élaboration modérées[Barton et al., 2005].
Les alcalins servent de fondants, c’est-à-dire qu’i ls permettent de diminuer de façon substantielle la viscosité, la température de transition vitreuse (Tg), ils augmentent la densité, l’indice de réfraction, le coefficient d’expansion thermique (CTE) et la conductivité électrique entre autres… L’addition d’un oxyde à la silice engendre le proce ssus de dissolution suivant. Les oxydes de métaux avec des cations possédant des degrés d’oxydation +1, +2, +3 peuvent être considérés comme existant sous la forme d’unités moléculaires de MO, MO et M O respectivement, ce qui conserve la stœchiométrie. L ’espèce moléculaire d’oxyde de métal peut réagir avec le réseau silicaté en convertissandes atomes d’oxygène pontants en atomes d’oxygène non-pontants. L’énergie de cette réactionentre en compétition avec l’énergie nécessaire pour garder les unités moléculaires d’oxyde métallique associé ou sous forme de clusters. En général, si le cation est un modificateur de réseau, de la sorte qu’il casse les liaisons Si-O-Si, le nombre maximum d’atomes d’oxyg ènes non-pontants qui peuvent se former est proportionnel à la valence du cation.
Les alcalins modifient donc la structure : chaque ion alcalin crée un nouvel NBO (Non- Bridging Oxygen ou Oxygène Non-Pontant) et chaque mole d’oxyde alcalin Na 2O produit deux atomes d’oxygène non-pontants (Figure 2).
Rôle des ions terres rares ?
Différents critères ont été créés afin de pouvoirlasserc les éléments suivant leur rôle au sein du réseau vitreux. Quelques uns sont brièvement présentés :
Zachariasen a, par exemple, proposé la notion de formateur et de modificateur de réseau. Pour lui, le formateur (silice par exemple) participe à la formation d’un réseau 3D désordonné tandis que les modificateurs rompent lesliaisons covalentes (alcalin et alcalino-terreux) [Zarzycki, 1991].
Dietzel classe les cations en fonction de leur force de champ pour leur attribuer un rôle. La force de champ A est alors le rapport de l a valence du cation sur la somme des rayons ioniques au carré [Vogel, 1985] : A Zc avec Zc= valence du cation ; rc : le rayon du (r r )2 cO cation (en Å) et r O le rayon ionique de l’oxygène (en Å).
Ainsi, pour des valeurs de A supérieures à 1,3, les cations sont plutôt formateurs, pour des valeurs inférieures à 0,4, ils sont modificateurs. Sinon, ils sont dits intermédiaires, c’est-à-dire que suivant la composition du verre, ils jouent un rôle de modificateur ou de formateur de réseau.
Stanworth, lui, prend en compte la liaison chimique et l’électronégativité des cations [Stanworth, 1971]. Pour lui un formateur se caractérise par une liaison fortement covalente car c’est l’architecture du réseau. Un modificateur aura une liaison plus ionique (moins covalente). Ce modèle se base sur les différences d’électronégativité de Pauling en estimant le degré d’ionicité de la liaison. Plus la différenced’électronégativité est petite, plus la liaison est covalente donc plus l’ion aura un caractère formateur de réseau.
Sun a établi une corrélation entre la force des liaisons et la capacité à vitrifier des oxydes. La force de la liaison M-O dans un oxyde est calculée à partir de l’énergie de dissociation de l’oxyde cristallin qui est divisée par sa coordinence. MxOy -> xM + yO (Energie de dissociation)
Par la suite, sont déterminés les formateurs de réseau qui possèdent une force de liaison supérieure à 80 kcal/mol, les modificateurs qui ont une force de liaison inférieure à 60 kcal/mol. Les intermédiaires se situent entre ces deux bornes.
Rawson a amélioré le critère de Sun en remarquant ueq pour relier la faculté de vitrifier à la possibilité de rompre des liaisons à la température de fusion, il faut tenir compte de la force de la liaison mais aussi de la quantitéd’énergie thermique disponible pour casser cette liaison. Cette quantité d’énergie est évaluéegrâce à la température de fusion. Le paramètre de Rawson correspond donc au critère de Sun divisé par la température de fusion de l’oxyde en kelvin.
Si tous ces critères ne sont pas parfaits (ils souffrent de nombreuses exceptions), ils permettent néanmoins d’avoir une idée du rôle des oxydes. L’ensemble de ces critères montrent notamment que les ions terres rares se situent entre les cations modificateurs (Na, Ca) et les cations intermédiaires (Al). Quelques critères sont montrés dans le Tableau 2.
Formation de verres avec terre rare
Dans des verres très simples, l’ajout d’ions de terre rare est plutôt délicat, notamment du fait de la force de champ relativement élevée des terres rares (Figure 15).
-Verres silicatés
La silice pure et les oxydes de terres rares possèdent des températures de fusion très élevées qui rendent l’élaboration de verres richesen terres rares très difficile. De plus, les terres rares sont difficilement solubles dans la silice comme le montre le diagramme de phase binaire proposé en Figure 14.
L’ajout de quelques ppm de terres rares conduit à l a formation de clusters [Sen et Stebbins, 1995], [Sen, 2000]. 300 ppm de Nd2O3 au sein de SiO2 suffisent à former des clusters de terres rares par exemple [Sen et Stebbins, 1995]. Cette clusterisation est notamment expliquée par la grande force de champ des ions terres rares et de leur tendance à vouloir être entourés d’atomes d’oxygène non-pontants.
Globalement les coordinences des ions terres rares dans les matrices de silice vitreuse sont très faibles. La structure de la silice vitreuse étant très rigide, la coordinence des ions terres rares n’est pas satisfaite, cela engendre la tendance pour les terres rares à former des clusters pour partager leurs oxygènes non-pontants (dynamique moléculaire) [Du et Cormack, 2005].
Pour les solubiliser, il donc est nécessaire d’incorporer des codopants (oxyde de phosphate ou d’aluminium) qui forment une couche de solvatation autour des ions terres rares (terres rares solubles dans ces oxydes). Ceci conduit à un effet de déclusterisation, et il est ainsi possible d’introduire plus de terres rares sans modifier les propriétés optiques de la terre rare au sein de sa matrice.
-Verres boratés
Peu de travaux existent sur les verres binaires borate- terres rares du fait de la préparation des verres qui est délicate : la régionde formation de verre est en effet très limitée.
Néanmoins, des études par spectroscopies infrarougeet Raman ont été effectuées sur ce type de verre [Terashima et al., 1997], [Chakraborty et al., 1984]. Elles portaient sur des verres dans la zone de composition Ln2O3-3B2O3 (Ln=La, Pr, Nd, Sm) car c’est uniquement dans cette région qu’il est possible de former des verres homogènes avec les ions terres rares. Cette composition correspond au métaborate de terre rare. La structure vitreuse proche de cette composition est similaire à l’homologue crist allin. Une représentation schématique du cristal métaborate de lanthanide (LnBO ) est montrée en Figure 16. Cette structure est constituée de chaînes d’unités trigonales BO et tétraédriques BO. Il y a deux fois plus d’unités trigonales que d’unités tétraédriques. Elles sont connectées grâce aux ions lanthanides (qui sont de coordinence 10). Les terres rares ont une incidence non négligeable sur la structure car les unités boroxols ne sont plus présentes ici.
Il est à noter que ce verre n’est pas réalisable avec tous les ions terres rares : les systèmes boratés avec des ions europium, gadoliniumou holmium (avec un petit rayon ionique) forment difficilement un verre et dévitrifient en orthoborate (LnBO3) [Chakraborty et al., 1985].
Il a par ailleurs été montré que lorsque la quantité de LaO est faible, l’ion lanthane s’insère entre les larges unités boratées et la structure locale de l’ion La 3+ est plutôt distordue. Lorsque le pourcentage de La2O3 augmente, l’ion lanthane est entouré par de plus petites unités anioniques et la structure locale de l’ion semble mieux organisée. L’ajout d’atome de lanthane provoque la rupture des chaînes boratées en de plus petites unités [Kajinami et al., 2006].
Structure des verres sodiques avec terres rares
-Dans les verres boratés : changement de coordinence des atomes de bore
L’ajout de modificateurs au sein des verres boratés facilite la formation de verres avec des oxydes de terres rares et élargit fortment le domaine de formation des verres.
L’ajout progressif de Nd 2O3 dans la matrice Na2B4O7 semble provoquer la diminution du nombre de groupements boroxols et donc la conversion de BO en unités BO [Culea et al., 2001]. Ce comportement suggère un caractère modificateur de réseau pour les ions néodymes III. Une autre étude, sur un système LaO -CaO-B O , montre que l’ajout de La O provoque une diminution de la proportion de BO4, ce qui implique une augmentation de la quantité des BO et la formation d’un réseau moins interconnecté[Dyamant et al., 2008]. Ce comportement caractérise également un rôle de modificateur de réseau selon Dyamantet al.
-Dans les verres silicatés : création d’oxygène non-pontant
L’ajout d’un cation au sein du réseau vitreux engendre des changements locaux au niveau de la distribution des atomes d’oxygène non-pontants et pontants [Bonamartini Corradi et al., 2005]. L’addition d’un ion modificateur augmente les qua ntités d’atomes d’oxygène non-pontants ce qui conduit à une dépolymérisation du réseau vitreux. Les atomes d’oxygène pontants et non-pontants peuvent être distingués et quantifiés de manière plus ou moins précise par XPS [Smets et Krol, 1984], Raman [Krol et Smets, 1984] et RMN de 17O [Lee et Stebbins, 2009]. Dans Na2O.SiO2, la dynamique moléculaire montre que l’ion lanthane se comporte comme un modificateur car il augmente la quantité d’atomes d’oxygène non-pontants [Park et al., 2002]. Toutefois, le choix des modèles de potentiel est un facteur déterminant pour l’issue de la dynamique moléculaire, l’utilisation de potentiels divers ne conduisant pas toujours au même résultat[Park et al., 2002].
Par ailleurs, l’environnement Na/NBO est plus flexible que le réseau silicaté, il permet ainsi à l’ion terre rare d’adopter sa configuration optimale.
Deux types d’introduction des ions terres rares sont particulièrement étudiés au sein des matrices silicatées : l’ajout des oxydes correspondants sur la composition globale ou la substitution de 3Na2O par 1La2O3 par exemple. Cette dernière substitution suppose qu’un ion La3+ est équivalent à trois ions Na+, c’est-à-dire que le comportement des ions La 3+ est assimilable à celui de trois Na + (même charge et même nombre d’oxygènes non-pontants).
En effet, pour la substitution de 3Na2O par 1La2O3, il est attendu, en théorie, un ratio NBO/T (rapport du nombre d’atomes d’oxygène non-pontants par tétraèdre de silicium) constant si l’ion lanthane (chargé 3+) se comporte comme trois ions sodium.
L’ajout d’oxyde de terre rare sur la composition to tale engendre la formation d’atomes d’oxygène non-pontants [Schaller et al., 1999]. En effet, les contributions associées aux unités Q deviennent plus importantes au fur et à mesure de l’augmentation de la concentration en oxyde de lanthane. De plus, une contribution est également associée à des Q3-La, c’est-à-dire un Q 3 où l’ion sodium est remplacé par un ion lanthane : cette contribution se distingue des Q3 ‘classiques’ car le remplacement de Na par La enge ndre une augmentation de la force de champ du cation et donc un décalage vers les plus petits nombres d’onde en spectroscopie Raman. En outre, la température de transition vitreuse (Tg) augmente même lorsque le ratio NBO/T augmente [Branda et al., 1981], [Buri et al., 1982], [Coon et Shelby, 1994]. Ce même comportement a déjà été observé avec CaOTout. ceci est dû à la plus grande force de la liaison La-O par rapport à la liaison N a-O [Branda et al., 1981], [Buri et al., 1982].
Expérimentalement, lors de l’ajout de 1La O au détriment de 3NaO, Schaller et al. [Schaller et al., 1999] observent en Raman l’apparition de nouvelles contributions pour des nombres d’onde plus faibles qui traduit l’existence d’unités Qn avec des n plus petits. En revanche, la RMN MAS.
Si dévoile par déconvolution une proportion d’entités Q qui devient plus importante au détriment des entités Q. Les spectres Raman et RMN MAS de 29Si de cette série verrière sont reproduits en Figure 17.
D’après les déconvolutions des spectres de RMN, Schaller en déduit que le ratio NBO/T diminue au fur et à mesure de la substitution de 3Na2O par 1La2O3. L’ion lanthane ne forme pas autant d’oxygènes non-pontants que trois ions sodium. Pour lui, deux scénarios plausibles peuvent conduire à ce résultat :
– soit une certaine quantité des atomes d’oxygène iésl aux Si et La ne se comporterait pas comme des oxygènes non-pontants mais plutôt comme des oxygènes pontants.
– soit que certains atomes d’oxygène liés à des ions lanthane ne soient pas liés à des atomes de silicium. Dans ce cas, une partie des La2O3 introduite est isolée dans le réseau silicaté : il y aurait alors des atomes d’oxygène dits libres. Les ions lanthane associés à ces oxygènes libres ne modifieraient pas le réseau, cequi expliquerait que le ratio NBO/T soit plus faible qu’attendu. Ceci traduirait l’existence d’hétérogénéités.
Interdiffusion / échange d’ions
L’eau réagit avec la surface des verres silicatés pour former une couche de silice hydratée. Si le verre contient des ions alcalins (comme Na+), l’hydratation est précédée par l’échange d’ions entre l’hydrogène (ou l’ion hydronium H3O+) provenant de l’eau et les ions alcalins du verre [Rana et Douglas, 1961 a], [Doremus, 1975]. Cette étape ne peut avoir lieu sur de la silice amorphe car cette dernière est exempte de cations modificateurs.
Les ions mobiles (alcalins, Na+ le plus souvent) du verre sont libérés du réseauilicaté,s la matrice globale restant intacte. Cette réaction est sélective puisqu’il y a seulement les cations modificateurs du réseau vitreux qui sont ainsi libérés du fait de leur faible force de liaison au sein du réseau. Ces cations s’échangentavec les protons de la solution (H+ ou H3O+) : c’est l’interdiffusion :
Si-O- Na+ + H2O => Si-OH + Na+ + OH-
Si-O-Na+ + H3O+ => Si-OH + Na+ + H2O
Cette réaction n’est pas favorisée en milieu basique car OH- est présent du côté droit de l’équation, en revanche elle est favorisée en milieu acide (H3O+). Cette réaction provoque une hausse du pH.
L’échange d’ions peut être associé à une constanted’équilibre K [Si OH ][Na ] qui [Si O Na ][H ] + fait intervenir le site d’échange (Si-O-Na ) et la distribution de charge électronique [Bunker, 1994]. Ces sites étant dépendants de leur nature, la composition du verre (et probablement la structure) est directement liée à l’interdiffusion.
Formation de produits d’altération
La formation de produits d’altération dépend de lacomposition du verre et comprend d’une part la formation d’une couche d’altération et/ou, d’autre part, la précipitation de phases secondaires. Elle se produit suite à l’augmentation des concentrations en solution des différentes espèces hydrolysées. La vitesse d’altération chute fortement et la dissolution est alors incongruente. Il y a rétroaction des élémentssolubilisés due soit à de l’adsorption, soit au redépôt des espèces en solution [Jantzen, 1992].
Deux mécanismes de formation de la couche d’altération sont proposés selon l’élément : l’une est la réaction de condensation uiq permet une recombinaison in situ des éléments les moins solubles (Si par exemple) ; l’autre est la précipitation de phases secondaires.
– Formation de la couche d’altération
Les éléments constitutifs du verre peuvent être classés dans trois groupes en fonction de leur comportement vis-à-vis de la couche d’altération [Godon et al., 2004] :
– mobiles, c’est-à-dire qu’ils ne sont pratiquement pas rete nus dans la couche d’altération, c’est le cas des alcalins (Na+, Li+) et du bore par exemple
– intermédiaires: ils sont alors partiellement retenus, c’est le cas notamment du calcium, du silicium et de l’aluminium
– retenus quasi-intégralement comme le zirconium, les terresrares, les actinides.
Globalement, la couche d’altération est majoritairement constituée de silice (cation majoritaire dans le verre initial). Il est important de noter que cette couche n’est pas le verre seulement appauvri en éléments mobiles. En effet, esd altérations de verres dans de l’eau enrichie en 17O ont été menées par Bunkeret al [Bunker et al., 1988]. Elles montrent clairement que le verre réagit avec l’eau car il est ainsi possible d’obtenir les signatures des différents sites de la couche d’altération par RMNde 17O (le verre étant non enrichi en17O).
Les groupements silanols (Si-OH créés précédemmentpar hydrolyse) présentent une forte tendance à se condenser les uns aux autres, r eformant des liaisons Si-O-Si et promouvant ainsi la repolymérisation du réseau de ellet sorte qu’il y ait un maximum de liaisons siloxanes (Si-O-Si) et un minimum de groupes Si-OH non condensés. La couche non cristalline ainsi formée est appelée gel. Ce gel est amorphe, poreux et hydraté.
Le mécanisme actuellement le plus probable pour la réaction de condensation induit l’attaque d’un silanol déprotoné (nucléophile) surune espèce silicatée neutre [Brinker, 1988] : =SiO- + Si(OH)4 =Si-O-Si= + OH-
La réaction ‘dépend’ du point isoélectrique de la ilices où la surface des silanols peut être déprotonée suivant leur acidité. L’acidité dessilanols dépend des autres substituants sur le silicium. La vitesse de condensation est maximale dans des solutions près des pH neutres où des quantités suffisantes de silanols protonés tedéprotonés existent. La vitesse minimale est observée près du point isoélectrique.
Le rapport de stage ou le pfe est un document d’analyse, de synthèse et d’évaluation de votre apprentissage, c’est pour cela chatpfe.com propose le téléchargement des modèles complet de projet de fin d’étude, rapport de stage, mémoire, pfe, thèse, pour connaître la méthodologie à avoir et savoir comment construire les parties d’un projet de fin d’étude.
|
Table des matières
CHAPITRE 1. S1. STRUCTURE ET ALTERATION DES VERRES
I. LA STRUCTURE DES VERRES SIMPLES
I.1. De la silice au verre borosilicaté
I.2. Les terres rares dans les verres
II. ALTERATION DES VERRES PAR L’EAU
II.1. Mécanismes chimiques d’altération
II.2. Cinétiques
II.3. De la silice au verre borosilicaté – effet de composition
II.4. Structure et morphologie des couches d’altération
III. CONCLUSION ET OBJECTIFS DE LA THESE
CHAHAPITRE 2. T2. TECHNIQUES ET PROTOCOLES EXPERIMENTAUX
I. COMPOSITIONS VERRIERES
II. PRINCIPALES TECHNIQUES DE CARACTERISATION
II.1. Résonance Magnétique Nucléaire (RMN)
II.2. Spectroscopie Raman
II.3. Spectroscopies de fluorescence
II.4. Secondary Ion Mass Spectrometry (SIMS)
II.5. SAXS (Small-Angle X-ray Scattering) et USAXS
II.6. Microscopie Electronique à Balayage (MEB)
II.7. Microscopie Electronique à Transmission (MET)
III. PROTOCOLES EXPERIMENTAUX
III.1. Elaboration et préparation des verres
III.2. Protocoles de Lixiviation des verres
CHAPITRE 3. I3. INFLUENCE DU LANTHANE SUR LA STRUCTURE DU RESEAU VITREUX
I. VUE D’ENSEMBLE DE LA MATRICE VITREUSE
I.1. Evolutions des vibrations des espèces silicatées
I.2. Evolutions des groupements boratés et borosilicatés
I.3. Evolution de la vibration La-O
I.4. Effet de l’oxyde de lanthane sur la structure globale du verre
II. CONNECTIVITE DES ATOMES DE SILICIUM
II.1. Spectres de RMN MAS de 29Si
II.2. Evolution de la largeur des spectres de RMN MAS de 29Si
II.3. Evolution du déplacement chimique de 29Si
II.4. Confrontation aux données de la littérature
II.5. Conclusion
III. COORDINENCE DES ATOMES DE BORE
III.1. Evolution de la coordinence des atomes de bore
III.2. Evolution du déplacement chimique du 11B
III.3. Comparaison Raman et RMN MAS de 11B
III.4. Comparaison à d’autres éléments
III.5. Conclusion
IV. ENVIRONNEMENT DE L’ION SODIUM
IV.1. Spectres de RMN MAS de 23Na
IV.2. Evolution du déplacement chimique de 23Na
V. ENVIRONNEMENT DE 17O
V.1. Identification des groupements 17O
V.2. Complexification du système : vers les borosilicates de terres rares
V.3. Présence d’oxygènes dits libres ?
VI. CONCLUSION
CHAPITRE 4. R4. ROLE DES TERRES RARES AU COURS DE L’ALTERATION
I. VITESSE INITIALE DE DISSOLUTION V0
I.1. Effet des oxydes de terres rares (nature et concentration)
I.2. A quoi peut être reliée la vitesse initiale de dissolution ?
I.3. Comparaison à d’autres éléments
I.4. Rôle des oxydes de terres rares sur la vitesse initiale d’altération
I.5. Que deviennent les ions terres rares ?
I.6. Conclusion sur l’effet des oxydes de terres rares sur la vitesse initiale de dissolution
II. ALTERATION EN MODE STATIQUE : LES IONS TERRES RARES ANALOGUES DES IONS CALCIUM OU ZIRCONIUM ?
II.1. Impact des oxydes de terres rares (nature et concentration) sur le pourcentage d’altération et la vitesse d’altération
II.2. Que deviennent les terres rares au cours de l’altération ?
II.3. Comment sont réparties les terres rares, quelles sont les modifications structurales ?
II.4. Quel est le rôle des ions terres rares au cours de l’altération ?
II.5. Conclusion sur l’effet des ions terres rares sur les altérations en mode statique
III. ROLE DES IONS TERRES RARES DURANT LES DIFFERENTS MODES D’ALTERATION
CHAPITRE 5. E5. EVOLUTION DE L’ENVIRONNEMENT LOCAL DES IONS TERRES RARES AU COURS DE L’ALTERATION
I. EUROPIUM TRIVALENT EN TANT QUE SONDE STRUCTURALE
I.1. Excitation continue
I.2. Déclins de luminescence
I.3. Effet de la température
II. ANALOGIE LANTHANE-EUROPIUM
II.1. L’ion europium III
II.2. Degré d’oxydation
III. INFLUENCE DE LA CONCENTRATION EN LANTHANE SUR LA LUMINESCENCE DE L’ION EUROPIUM TRIVALENT
III.1. Evolution de la largeur à mi-hauteur des transitions 5D07FJ
III.2. Evolution du paramètre d’asymétrie
III.3. Détermination du nombre de sites distincts
IV. DETERMINATION DES ENVIRONNEMENTS DE L’ION EUROPIUM TRIVALENT
IV.1. Environnements de l’ion europium trivalent au sein des verres borosilicatés sains
IV.2. Comparaison des environnements de l’ion europium trivalent à une référence vitreuse silicatée
V. EVOLUTION DES ENVIRONNEMENTS DE L’ION EUROPIUM TRIVALENT AU SEIN DES VERRES ALTERES
V.1. Evolution globale des spectres en excitation continue
V.2. Evolution du paramètre d’asymétrie
V.3. Déclins de luminescence
V.4. Excitation sélective
VI. CONCLUSION
CONCLUSION GENERALE
REFERENCES BIBLIOGRAPHIQUES
Télécharger le rapport complet