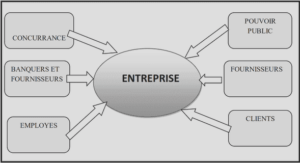
Télécharger le fichier pdf d’un mémoire de fin d’études
Changements et mise en place de la nouvelle réglementation
Pourquoi une nouvelle réglementation ?
Tout d’abord, il nous faut faire un retour en arrière pour définir la précédente réglementation européenne. Ainsi, la Directive Européenne 93/42/CEE est un texte réglementaire rédigé par l’Union Européenne, appliqué depuis 1993, donnant des indications communes concernant la mise sur le marché des dispositifs médicaux. Cette directive, applicable à l’ensemble des états membres de l’Union Européenne, a été enrichie par d’autres directives depuis près de 25 ans. Chacune de ces directives ont été transposées sur la législation de chaque membre de l’Union Européenne. Cela laisse ainsi libre cours à l’interprétation de chaque état membre, concernant les obligations de mise sur le marché des dispositifs médicaux. De plus, depuis de nombreuses années, le secteur des dispositifs médicaux était agité par des scandales massivement médiatisés tels que l’affaire des prothèses mammaires Poly Implant Prothèse (PIP) ou encore les prothèses de hanche à couple de frottement métal – métal. La capacité de la directive à assurer la sécurité des patients a ainsi été remise en cause. Pour remédier à ces disfonctionnements, et aux différents scandales du monde du dispositif médical, le parlement Européen a voté en Avril 2017 la mise en place d’un nouveau texte visant à unifier l’ensemble des acteurs du dispositif médical sous un seul et même règlement, plus complet suivant le contexte technologique actuel. Ce nouveau règlement a pour but d’améliorer la traçabilité et la transparence au niveau Européen, mais aussi de pouvoir surveiller de façon plus accrue les organismes notifiés. Le règlement sur les dispositifs médicaux est entré en vigueur après sa publication au Journal Officiel de l’Union Européenne le 5 mai 2017, et comme nous l’avons vu précédemment, sera appliqué à compter du 26 mai 2020. Cette transition doit être appliquée par les entreprises mais également par les organismes notifiés ainsi que les différents acteurs du secteur concernés par le cycle de vie des DM et des DMDIV comme nous allons le voit par la suite 24.
Principaux changements
La réglementation 2017/745 a apporté de nombreux changements comme nous avons pu voir le voir en introduction. De façon plus détaillée, nous pouvons citer 26 :
• Une modification importante des procédures de l’évaluation de la conformité, et en particulier des procédures d’évaluation clinique pour toutes les catégories de produits avec une procédure particulière pour les plus innovants
• Un renforcement des procédures de vigilance pour permettre une meilleure détection des signaux faibles
• Un renforcement de la transparence des produits (création d’une base de données européenne) et de la traçabilité (identifiant unique : UDI)
• Un élargissement du champ des dispositifs médicaux.
• Un renforcement du rôle des opérateurs économiques (distributeurs, importateurs, etc.)
• L’obligation d’avoir une personne chargée de veiller au respect de la réglementation.
Ces modifications permettront alors d’apporter les améliorations suivantes 31 :
• Un contingent de définitions enrichi et aligné avec les autres textes européens.
• Des règles claires pour les investigations cliniques.
• Un format de documentation technique intelligent. S’inspirant du format STED (Summary Technical Documentation) établi par le GHTF (Global Harmonization Task Force), cette architecture harmonisée facilitera les enregistrements à l’international, diminuant ainsi les coûts associés • Un Identifiant Unique des Dispositifs facilitant la gestion des stocks, la traçabilité (distribution et utilisation), la lutte contre la falsification…
• Une implication de tous les opérateurs économiques de la chaîne avec la responsabilisation des distributeurs.
• La prise en compte des nanomatériaux, des logiciels, des services localisés hors Europe.
• De la transparence avec la création de EUDAMED.
• Un renforcement des règles de désignation et de surveillance des organismes notifiés.
Ainsi, plusieurs finalités sont mises en évidence 8 :
• L’amélioration de la qualité, de la sécurité et de la fiabilité des dispositifs médicaux.
• Une plus grande transparence des informations pour les consommateurs.
• Un renforcement de la vigilance et de la surveillance du marché L’un de ces nouveaux points nécessitant un focus particulier est l’élargissement du champ d’application des dispositifs médicaux afin notamment d’intégrer dans la définition des dispositifs médicaux tous les dispositifs destinés à être utilisés à des fins de prévision et de pronostic d’une maladie. Ainsi, concernant les logiciels, un logiciel destiné aux fins médicales exposées à cet article est qualifié de dispositif médical. Seuls les logiciels destinés à des usages généraux, en particulier liés au mode de vie ou au bien-être, n’entrent pas dans le champ d’application de la réglementation relative aux dispositifs médicaux. Toutefois, certains produits, pour lesquels seule la fonction esthétique ou non médicale est mise en avant, peuvent relever du règlement européen lorsqu’ils sont semblables à des dispositifs médicaux. En effet, conformément à l’article 1 du règlement relatif aux dispositifs médicaux, des spécifications communes seront appliquées aux dispositifs n’ayant pas de fonction médicale mais qui figurent sur une liste établie en annexe du règlement (ex. équipements émettant des rayonnements électromagnétiques à haute intensité tels que les équipements à lumière pulsée utilisés pour la suppression des tatouages ou l’épilation). Les dispositifs qui ont à la fois une destination médicale et non médicale sont également mentionnés comme faisant partie du champ d’application du règlement 7. Cependant, ces différents points positifs sont à nuancer. En effet, le texte n’est pas parfait selon certains professionnels de santé impliqués dans le secteur des dispositifs médicaux qui expliquent que cette nouvelle réglementation contient encore des lacunes et que certains points n’ont pas été traités : quid de la résolution de conflits entre les acteurs (type médiation) dont l’efficacité a été démontrée par la FDA ? Ces professionnels mettent en avant que ce n’est pas la réglementation qui pose problème mais plutôt la façon dont la société l’utilise. Le réel point noir, c’est l’engagement des industriels dans ce changement. Lutter contre le changement ou le nier, c’est permettre des lectures abusives dont les professionnels de santé et les patients paieront le prix à terme avec la disparition de produits qui ont un réel bénéfice pour la santé et une diminution de l’arrivée sur le marché d’innovations majeures.
Ainsi, le Règlement n’est pas considéré comme un désastre, mais il peut le devenir, selon certains professionnels de santé, si les fabricants ne prennent pas dès maintenant la transition en main avec une vision stratégique et proactive de la situation. Cela nécessite une implication des dirigeants et une collaboration entre industriels pour faire valoir des solutions de bon sens plutôt que des solutions de technocrates.
Les acteurs de la nouvelle réglementation
En plus de l’élargissement de son champs d’application sur les dispositifs médicaux ainsi que la modification de leur classification, la nouvelle règlementation définit de nouveaux acteurs ainsi que de nouvelles responsabilités incombant aux acteurs de la réglementation déjà établis, tel que les Organismes Notifiés ou les Autorités Compétentes. Tout cela en prenant également en compte les responsabilités nouvelles ou accrues des opérateurs économiques 8 :
• L’autorité compétente : Elle est définie comme une émanation de l’Etat chargée notamment de la surveillance du marché national des dispositifs médicaux. Au-delà de son devoir de surveillance et de nomination des organismes notifiés, elle dispose de la prérogative de sanction auprès des fabricants. Mais elle peut également auditer périodiquement l’organisme notifié et apprécie les certificats qu’il délivre. En France, l’autorité compétente est l’Agence Nationale de Sécurité du Médicament et des produits de santé (ANSM). Le règlement confirme le rôle des autorités compétentes avec un niveau de précision plus important dans sa description notamment en matière de vigilance. En outre, les autorités compétentes doivent désormais coopérer entre elles et avec la Commission Européenne en échangeant régulièrement des informations afin de faciliter l’application uniforme du Règlement.
• Le groupe de coordination : Afin de correspondre à l’esprit du Règlement, à savoir mettre l’accent sur la sécurité des utilisateurs, la Commission a prévu que la nomination des organismes notifiés serait également analysée par un nouveau groupe – le Groupe de Coordination en matière de Dispositifs Médicaux (GCDM) – qui a aussi pour compétence d’auditer les organismes notifiés. Cet organe est également un relai de communication entre les organismes notifiés et la Commission dans la mesure où le GCDM sera au centre d’un processus de reporting applicable aux dispositifs de classe III. Le GCDM a ainsi pour mission d’assurer la surveillance du marché et le respect des procédures.
• Les organismes notifiés : Désignés par l’autorité compétente de chaque Etat, les organismes notifiés assurent l’interface entre le fabricant et l’autorité compétente. Ses principales missions d’instruction, de contrôle et de vérification sont accompagnées d’une obligation d’indépendance, d’intégrité, d’impartialité, de formation et de compétence. L’organisme notifié est investi de la mission d’évaluation de la conformité d’un DM avant sa mise sur le marché. Pour mener à bien sa mission principale, il dispose du droit de diligenter des audits auprès des fabricants. Avec les fabricants, ce sont les acteurs les plus touchés par les modifications du Règlement. En effet, par le biais de l’Annexe VII relative à la procédure uniformisée à suivre afin d’obtenir et de conserver le statut d’organisme notifié, les organismes notifiés doivent être réévalués pour acquérir ce statut conformément à la nouvelle réglementation sur les dispositifs médicaux, même si l’organisme notifié possédait déjà ce statut conformément aux Directives actuelles. Une nouvelle procédure prévoit également que les organismes notifiés informent la Commission Européenne des demandes qu’ils reçoivent concernant les dispositifs de la classe III (DM) ou classe D (DMDIV). Ces dossiers pouvant donner lieu à un examen par le Groupe de Coordination en matière de Dispositifs Médicaux Chaque Etat membre doit désigner une autorité chargée d’évaluer les organismes notifiés (En France, le LNE/G-MED est le seul organisme notifié français au titre des directives européennes applicables aux dispositifs médicaux). Cette autorité est tenue d’évaluer les organismes notifiés, filiales et sous-traitants compris, au moins une fois par an, et procède à une évaluation complète de l’organisme trois ans après la notification, puis tous les quatre ans. De plus, par l’intermédiaire du Groupe de Coordination en matière de Dispositifs Médicaux (GCDM), la Commission participe désormais à la désignation des organismes notifiés 7 : • La Commission et le GCDM désignent une équipe d’évaluation de l’organisme ; • Le GCDM émet une recommandation sur le projet de désignation. L’autorité doit tenir compte de cette recommandation lorsqu’elle statue sur la désignation de l’organisme notifié .
• L’équipe d’évaluation participe à l’évaluation complète de l’organisme notifié qui doit être réalisée trois ans après notification, puis tous les quatre ans. Cependant, il existe une volonté européenne de diminuer le nombre d’organismes notifiés (-40% en 5 ans) : Cela a accru fortement la charge de travail des organismes toujours en place, qui n’acceptent pas toujours de nouveaux dossiers, ou alors avec des délais pouvant aller de 6 mois à 1 an. Par ailleurs, les organismes voient leur activité très largement augmentée par 27 : • L’augmentation très forte, recommandée par la réglementation, des audits inopinés, conduisant parfois à des situations complètement incongrues pour les sous-traitants (succession d’audits inopinés, parfois réalisés par le même organisme qui certifie le sous-traitant, parfois 2 audits inopinés en quelques mois par le même organisme et sur le même produit, …) 27.
• L’obligation pour l’ensemble des organismes à être notifiés par leur autorité compétente avant fin juillet 2019 (alors que les fabricants devront être conformes au nouveau règlement moins d’un an plus tard, le 26 mai 2020). Ces délais extrêmement tendus risquent de peser encore plus sur les relations organismes/fabricants/sous-traitants. Restrictions des compétences de certains organismes notifiés : les fabricants devront faire appel à plusieurs organismes notifiés, car celui qui est compétent en neurologie ne le sera pas forcément sur le produit que le fabricant souhaite développer pour l’orthopédie par exemple. Tous ces changements auront pour impact de freiner le développement des fabricants et de les niveler, car les petites entreprises (qui représentent 90 % de la filière en Bourgogne Franche Comté par exemple) seront vite évincées par manque de moyens… avec pour conséquence directe le frein porté à l’innovation 27.
Il faut également traiter du cas particulier des OBL (Own Brand Labeller) : Il s’agit d’un opérateur économique qui achète un dispositif médical marqué CE à un fabricant « d’origine » et qui le met sur le marché avec les mêmes revendications et sans aucune modification sous sa propre marque. Dans le cadre des Directives, l’OBL n’était pas défini au sein des textes, seul le fabricant l’était. Toutefois, il était considéré comme tel, et cette interprétation avait été confirmée par une recommandation de la Commission Européenne de 2013. Le nouveau règlement clarifie la définition de cet opérateur économique, bien que le terme « OBL » n’apparaisse pas dans le texte. En vertu de ses missions et du texte du Règlement, l’OBL est donc désormais un fabricant au sens règlementaire du terme, et à ce titre, il doit être en possession de l’ensemble du dossier technique et doit le tenir à disposition des autorités compétentes. Toutefois, il lui est désormais possible, dans les cas où il ne peut disposer du dossier technique, (si par exemple son concurrent ne souhaite pas lui fournir les éléments), d’avoir un accord avec cette entreprise lui demandant de faire apparaître le nom de marque de son produit dans le dossier technique et sur sa déclaration de conformité. Cela permettra alors à l’OBL, par le biais de cet accord, de ne plus être défini comme un fabricant, mais comme un distributeur, et de voir dans le cadre de la réglementation ses obligations allégées 8.
Développement et données cliniques
Nous allons dans un premier temps faire un point sur la nécessité des données cliniques dans le développement et la mise sur le marché des dispositifs médicaux. En effet, pour être mis sur le marché, un dispositif médical doit satisfaire aux exigences essentielles des directives européennes qui lui sont applicables en tenant compte de sa destination. Pour la directive 93/42/CEE relative aux dispositifs médicaux, par exemple, l’exigence essentielle générale de l’annexe I mentionne au chapitre I.6 bis que « la démonstration de la conformité aux exigences essentielles doit inclure une évaluation clinique conformément à l’annexe X ». Cette dernière étant entièrement dédiée à l’évaluation clinique. L’évaluation clinique repose donc sur 18 : • Des données cliniques • Des investigations cliniques • Un suivi après mise sur le marché Il est à noter qu’un chapitre dédié à l’évaluation clinique est obligatoire dans la documentation technique de chaque dispositif médical. L’annexe X de la directive 93/42/CEE était d’ailleurs dédiée à l’évaluation clinique. Elle décrivait les dispositions générales applicables en la matière, ainsi que les objectifs, les considérations éthiques et les méthodes d’investigations cliniques. Il était spécifié que les caractéristiques et performances d’un dispositif ainsi que l’évaluation des effets indésirables et du caractère acceptable du rapport bénéfice/risque doivent être fondés sur des données cliniques. L’annexe X de cette directive précisait également que tous les événements indésirables graves devaient être intégralement enregistrés et communiqués immédiatement à l’ensemble des autorités compétentes des Etats membres dans lesquels étaient réalisées les investigations cliniques 18. De plus, la directive 2007/47/CE avait précisé la directive 93/42/CEE quant à la définition de « données cliniques ». L’article 1.1.k mentionnait ainsi que les données cliniques étaient des informations relatives à la sécurité et aux performances obtenues dans le cadre de l’utilisation clinique d’un dispositif 18. Après ces précisions sur l’ancienne réglementation, nous pouvons dire que les données cliniques peuvent provenir : – Des investigation(s) clinique(s) du dispositif concerné, ou – Des investigation(s) clinique(s) ou d’autres études citées dans la littérature scientifique d’un dispositif similaire pour lequel l’équivalence avec le dispositif concerné peut être démontrée, ou – Des rapports, publiés ou non, relatifs à une autre expérience clinique acquise sur le dispositif concerné ou un dispositif similaire pour lequel l’équivalence avec le dispositif concerné peut être démontrée. Nous pouvons donc établir que les principaux objectifs des investigations cliniques sont : • De vérifier que, dans des conditions normales d’utilisation, les performances du dispositif sont conformes à celles qui leur sont assignées. • De déterminer les éventuels effets secondaires indésirables dans des conditions normales d’utilisation et d’évaluer si ceux-ci constituent des risques au regard des performances assignées au dispositif. Nous avons pu constater que les sources des données cliniques peuvent être multiples. Mais elles peuvent également s’appuyer sur l’équivalence à un autre produit, à condition qu’une démonstration de cette équivalence soit faite et que celle-ci ne repose pas uniquement sur les caractéristiques techniques. Pour cela, le recours à la littérature est nécessaire pour renseigner cette l’évaluation clinique et permettre la démonstration de l’équivalence. Ainsi, l’évaluation critique de la littérature scientifique pertinente actuellement disponible concernant la sécurité, les performances, les caractéristiques de conception et de la destination du dispositif doit démontrer l’équivalence du dispositif avec le dispositif auquel se rapportent les données. A ce propos, il est essentiel de préciser que la démonstration de l’équivalence est une nouveauté du règlement 2017/745, en effet, l’accès aux données du dispositif équivalent (à sa documentation technique) doit être prouvé. Pour les classes III et DMI cela passera nécessairement par un contrat entre les fabricants, le plus souvent concurrents 10.
Il faut savoir que le recours à une équivalence est la solution la plus simple – c’est même le principe du 510k de la FDA – mais elle est réservée aux dispositifs non-innovants.
En contrepartie, l’investigation clinique est la voie la plus difficile car longue, risquée et chère (des centaines de k€). Elle est néanmoins obligatoire pour tous les dispositifs de classe III et implantables, sauf cas particuliers, parmi lesquels nous pouvons citer : les DM ayant déjà un marquage CE selon la directive, la modification d’un dispositif ayant un marquage CE, certains DM implantables comme les agrafes ou les appareils d’orthodontie, et si l’équivalence avec un autre dispositif peut être démontrée sur la base du dossier technique complet du DM équivalent 10. Une donnée importante à préciser est qu’il est nécessaire, dans la nouvelle réglementation, de choisir, pour les classes III et certains IIb, un groupe d’experts européens à consulter en amont de l’évaluation clinique.
Il faut savoir que le recours à un évaluateur indépendant sera :
• Systématique pour un DM dangereux et innovant.
• Souhaitable pour un DM maitrisé mais dont le contexte évolue fortement, par exemple en cas de matériovigilance récente dans le domaine.
• Dispensable pour un dispositif classe I en contexte stable. Ces dispositions précisent les modalités de coordination entre les états membres. Ainsi, si un Etat membre refuse ou interrompt une investigation clinique, il communique sa décision, ainsi que les raisons qui l’ont motivée, à tous les Etats membres et à la Commission européenne. Si un Etat membre a demandé une modification substantielle ou l’interruption provisoire d’une investigation clinique, il informe les Etats membres concernés des actions qu’il a engagées et des raisons qui les ont motivées. Il faut ensuite indiquer que le nouveau règlement s’est inspiré du guide MEDDEV 2.7/1 sur l’évaluation clinique des dispositifs médicaux. Ce guide européen MEDDEV 2.7.1 relatif à l’évaluation clinique, établi à l’attention des fabricants et organismes notifiés, constitue un document de référence comportant des informations très utiles sur l’analyse des données cliniques, sur le rapport d’évaluation clinique, sur la notion d’équivalence par exemple. Cependant, quelques modifications sont à noter : • Etablir un SCAC (Surveillance Clinique Après Commercialisation) qui est un plan de surveillance accompagné d’un rapport (avec mise à jour annuelle) pour les dispositifs médicaux implantables et dispositifs médicaux de classe III. • Constituer un RCSPC (Résumé des Caractéristiques de Sécurité et de Performances Cliniques) pour les dispositifs médicaux implantables et de classe III. • Rédiger le PSUR (Rapport périodique d’activité de sécurité) :
• Pour les dispositifs médicaux de classe IIa, IIb et III.
• Synthèse des résultats et des conclusions de l’analyse des données de surveillance
• Mise à jour :
– IIa : selon les besoins, au moins tous les 2 ans.
– IIb : au moins une fois par an.
– III : au moins une fois par an.
Pour terminer, nous allons maintenant apporter des précisions sur le cas des DM implantables et des DM de classe III : Dans le cas de dispositifs implantables et de dispositifs faisant partie de la classe III, les investigations cliniques doivent être réalisées, sauf si le recours aux données cliniques existantes peut être dûment justifié. Cette justification ne peut pas reposer uniquement sur l’équivalence à un autre DM. L’ensemble des informations relatives aux données cliniques sont à renseigner dans la base de données sur les dispositifs médicaux EUDAMED accessibles à l’ensemble des États membres.
Démarches renforcées
En effet, le règlement ne sera plus transposé dans le code de santé de chaque pays de l’Union Européenne mais devra être appliqué en l’état. Cela permettra d’éviter les disparités entre les différents organismes notifiés et états membres au niveau des exigences à respecter. Toujours dans un souci de rigueur uniformisée, le règlement introduit également des normes plus strictes pour les organismes notificateurs, notamment pour les dispositifs à haut risque à l’image des implants, des remplacements articulaires ou encore des pompes à insuline qui subiront une expertise complémentaire 24. Nous allons donc voir au travers de plusieurs points de quelle façon la nouvelle réglementation a renforcé ces démarches :
Documents qualités et techniques
Dans le cadre du durcissement des règles de classification, de plus en plus de dispositifs médicaux sont susceptibles d’être soumis aux règles de procédures d’évaluation de la conformité de l’Annexe IX relative au Système de Management de la Qualité. Le Système de Management de la Qualité est défini dans le cadre du règlement au sein de l’article 10.9. Il devra, pour être conforme, contenir plusieurs aspects définis au sein de l’article, parmi lesquels la proposition d’une stratégie de respect de la réglementation, l’identification des exigences générales en matière de sécurité et de performances et la recherche de solutions pour les respecter, la définition des responsabilités de la direction, la gestion des ressources, la gestion des risques sur l’ensemble du cycle de vie du dispositif, l’évaluation clinique et le suivi clinique après commercialisation… Le fabricant fait certifier son système par un organisme notifié, par le biais d’une demande faite à ce dernier et soumet son système de gestion de qualité à un audit pour déterminer son respect des exigences réglementaires. Le Règlement sur ce point, s’il présente des exigences élevées pour le fabricant, ne présente pas de bouleversement majeur. En effet, une partie de ces exigences était déjà traduite au sein de la norme ISO 13485. Cette norme énonce les exigences relatives au système de management de la qualité lorsqu’un organisme doit démontrer son aptitude à fournir régulièrement des dispositifs médicaux et des services associés conformes aux exigences des clients et aux exigences réglementaires applicables. Ces organismes peuvent être impliqués dans une ou plusieurs étapes du cycle de vie incluant la conception et le développement, la production, le stockage et la distribution, l’installation ou les prestations associées d’un dispositif médical. Les dernières évolutions de cette norme, par le biais de la norme ISO 13485/2016 se rapprochaient déjà des exigences du Règlement11. De plus, la documentation technique que le fabricant doit établir est précisée et détaillée à l’annexe II du règlement européen, notamment en ce qui concerne les éléments suivants 7 :
• Description et spécification du dispositif, y compris ses variantes et ses accessoires. Les éléments fonctionnels clés, tels que les logiciels y sont notamment précisés .
• Informations relatives aux étiquettes et notices d’utilisation des dispositifs médicaux .
• Informations sur la conception et la fabrication .
• Exigences générales en matière de sécurité et de performance .
• Analyse bénéfice-risque et gestion des risques .
• Vérification et validation du produit (résultats et analyses des études et essais, etc.).
Il apparaît ainsi que la majeure partie des exigences à rattraper se situeront sur le volet de la Surveillance Clinique Après Commercialisation, définie notamment à l’Annexe III 8. L’un des impacts les plus significatifs à ce titre sera l’apparition d’un nouvel acteur chez le fabricant, à savoir le responsable des affaires règlementaires.
Le chargé d’affaires réglementaires :
Issu à l’origine du monde de l’industrie pharmaceutique, le concept de « Qualified Person » a subi plusieurs évolutions au cours des itérations de la Nouvelle Approche avant d’être formalisé dans les termes de l’article 15 du Règlement. Cet article définit aussi bien les conditions d’exercice, les qualifications nécessaires, mais également les situations dans lesquelles la nomination du Responsable ne sera pas obligatoire. Le responsable aux affaires réglementaires devra pour être reconnu avoir un diplôme, un certificat ou un autre document de certification formelle sanctionnant des études universitaires en droit, en médecine, en pharmacie, en ingénierie ou dans une autre discipline scientifique pertinente, ou un cycle de cours reconnu équivalent par l’État membre concerné, et une expérience professionnelle d’au moins un an dans le domaine de la réglementation ou des systèmes de gestion de la qualité en rapport avec les dispositifs médicaux. Le responsable aux affaires réglementaires a ainsi à sa charge 8 :
• Le contrôle de la conformité des dispositifs au système de gestion de la qualité .
• La rédaction de la documentation technique et la déclaration de conformité UE ainsi que leur mise à jour .
• La vérification des obligations en matière de surveillance après commercialisation .
• La prise en charge des notifications dans le cadre des obligations de vigilance .
• Dans les cas d’une investigation, la fourniture de la déclaration selon laquelle le dispositif en question est conforme aux exigences générales en matière de sécurité et de performances indépendamment des aspects relevant de l’investigation clinique. Il est à noter que ces compétences peuvent être réparties entre plusieurs personnes à partir du moment où les périmètres de responsabilité respectifs sont clairement documentés. Il faut également préciser que les micros et petites entreprises, c’est-à-dire les entreprises de moins de 50 personnes et dont le chiffre d’affaires annuel ou le total du bilan annuel n’excède pas 10 millions d’euros ne seront pas soumises à cette obligation et il leur sera possible d’externaliser, par le biais d’une prestation de services, cette obligation 8.
Surveillance après commercialisation
La réglementation impose la libre circulation des DM dans toute la communauté européenne (CEE), chaque Etat membre assurant la sécurité sur son territoire 14.
La sécurité des dispositifs médicaux (DM) impose le dépistage de tout dysfonctionnement, il s’agit de la matériovigilance. Cette dernière repose sur la déclaration obligatoire d’incidents survenant autour des DM. A l’échelon local, elle est organisée autour de correspondants locaux chargés de déclarer les événements à l’ANSM (Agence nationale de sécurité du médicament et des produits de santé), de mener l’enquête initiale et de prendre les mesures conservatoires éventuelles. A l’échelon national, l’ANSM a pour mission d’évaluer la sécurité d’emploi, l’efficacité et la qualité des produits médicaux. Elle est chargée de centraliser, d’évaluer les déclarations de matériovigilance et de prendre les décisions de police sanitaire qui s’imposent (retrait, rappel, alerte). A l’échelon européen, les états membres échangent des informations de matériovigilance à l’aide d’une base de données commune. La Sofcot est d’ailleurs très vigilante sur ce sujet et a créé une commission matériovigilance14. Ainsi, le Règlement impose aux fabricants l’établissement d’un plan de surveillance après la commercialisation des DM. Celui-ci devra être mis en place pour chaque dispositif, en fonction de sa classe et de son type. Il précisera les méthodes et procédures qui seront mises à oeuvre pour permettre d’actualiser l’évaluation clinique du DM et les éventuelles contre-indications observées dans l’objectif de mettre à jour le rapport bénéfice/risque 25. A l’issue de l’application de ce plan, le fabricant sera tenu de rédiger des rapports de surveillance post-commercialisation synthétisant les résultats obtenus et les conclusions tirées de leur analyse ainsi que toutes les actions correctives et préventives éventuellement prises et leur justification. Pour les DM de classe IIa, IIb et III, le fabricant aura l’obligation de rédiger un « rapport périodique actualisé de sécurité », rapport plus complet précisant les principales constatations dans le cadre du suivi clinique post-commercialisation, le volume des ventes, l’estimation des populations utilisatrices concernées ainsi que la fréquence d’utilisation du DM. Ce plan devra être mis à jour au moins une fois par an, ou tous les deux ans pour les seuls DM de classe IIa 25. De plus, les fabricants de dispositifs de classe I sont également tenus d’élaborer un rapport de surveillance après commercialisation. Ces nouvelles mesures impliquent que les fabricants mettent en place des méthodes efficaces de surveillance de leurs dispositifs médicaux 7.
Enjeux pour les entreprises
Une étude réalisée par le SNITEM (Syndicat National de l’Industrie des Technologies Médicales) en 2017 met en évidence les enjeux qui se présentent au secteur du dispositif médical, dont les 3 principaux sont, selon eux 27 :
• Les exigences réglementaires croissantes :
o « L’obtention du marquage CE s’est vu conditionnée au cours des 15 dernières années à des exigences réglementaires croissantes et une « marche » sans précédent doit de nouveau être franchie par les entreprises avec la très récente adoption du nouveau règlement européen sur les dispositifs médicaux » remarque Éric Le Roy, Directeur Général du SNITEM. « Cela doit constituer un point d’attention majeur pour les pouvoirs publics ».
o Ressources humaines : les nouvelles exigences réglementaires impactent à la fois les entreprises et les organismes notifiés qui doivent renforcer leurs équipes avec des employés/auditeurs plus nombreux et mieux formés. Cela crée aujourd’hui une énorme tension sur le marché des emplois réglementaires, avec de très grosses difficultés de recrutements, et des salaires qui explosent dans le secteur privé.
• Les difficultés d’accès au marché : En dépit d’un bon soutien amont à l’innovation, la spécificité des dispositifs médicaux (petites séries de produits et multiples référencements) demeure insuffisamment prise en compte, notamment dans les appels à projets. « Le modèle d’innovation qui repose largement sur des apports graduels aux utilisateurs (patients, professionnels de santé) manque de reconnaissance et de valorisation. Les produits subissent des baisses de prix régulières sans que puisse être pris en compte l’impact sur les coûts de production » souligne Stéphane Regnault, Président du SNITEM.
• Un manque de financement post-amorçage : Les besoins en capitaux des entreprises augmentent en raison des exigences (réglementaire, R&D ou export) de plus en plus marquées. Plus de la moitié des entreprises interrogées évoquent le manque de financement post-amorçage comme un vrai déficit de compétitivité. Face à ces difficultés, l’export est un relais de croissance pour les entreprises de taille moyenne ou intermédiaire, qui gagneraient à chasser davantage « en meute » et à renforcer l’effet vitrine France 27.
Développement du marché
En France, on ne peut commercialiser un dispositif médical que s’il a fait l’objet d’une procédure de marquage CE. Ce sésame est la clef d’entrée sur le marché. C’est aussi une condition nécessaire mais pas suffisante pour obtenir une prise en charge par l’Assurance Maladie. Une fois marqué CE, un dispositif médical dispose de plusieurs voies de financement 38.
Marquage CE
Pour être commercialisés sur le marché européen, les dispositifs médicaux doivent être conformes aux exigences de sécurité et de performance définies par la réglementation européenne.
Cependant, un marquage CE médical n’est pas délivré ad vitam aeternam. En effet, l’ANSM, surveille les dispositifs en circulation. Parallèlement, les fabricants mettent en place un système de veille intégrant plusieurs critères. Dès la mise sur le marché des dispositifs médicaux, les autorités compétentes (en France l’ANSM) qui ont un pouvoir de police sanitaire, les surveillent à travers leurs activités de vigilance, de contrôle et d’inspection. En parallèle, pour les dispositifs de classe IIa, IIb, III, l’organisme notifié vérifie au moins tous les ans que les exigences réglementaires continuent à être remplies, avec un contrôle approfondi pour renouveler les certificats au terme de la période38.
Enfin, pour toutes les classes de dispositifs, les fabricants ont l’obligation de mettre en place un système de surveillance après commercialisation c’est-à-dire de collecter de façon proactive les données pertinentes sur la qualité, les performances et la sécurité d’un dispositif pendant toute sa durée de vie, afin de définir et d’appliquer toute mesure préventive ou corrective et d’en assurer le suivi. Ils doivent également mettre en place un processus réactif de traitement des incidents graves mettant en cause les dispositifs médicaux (matériovigilance) afin notamment de les signaler immédiatement aux autorités compétentes (en France, l’ANSM) 38.
L’évaluation clinique fait également partie des éléments essentiels de l’évaluation de la conformité des dispositifs médicaux.
Il est important de noter que le règlement européen 2017/745 sera d’application obligatoire le 26 mai 2020 et les directives 93/42/ CEE et 90/385/CEE seront abrogées à l’exception de quelques dispositions. Une période de transition sera alors instaurée jusqu’à cette date, permettant ainsi aux fabricants de choisir une procédure de marquage CE selon les directives 93/42/CEE et 90/385/CEE ou le règlement 2017/745. Les certificats délivrés au titre des directives par un organisme notifié resteront valables jusqu’à la fin de leur période de validité, au maximum 5 ans après leur délivrance et au plus tard le 27 mai 2024 sous réserve que les produits ne fassent l’objet d’aucune modification substantielle. Les dispositifs pourront continuer à être dans le circuit de distribution jusqu’au 27 mai 2024 6.
|
Table des matières
I. Introduction
II. Changements et mise en place de la nouvelle réglementation
A. Pourquoi une nouvelle réglementation ?
B. Principaux changements
C. Les acteurs de la nouvelle réglementation
D. Planning de mise en place du changement
III. Exigences réglementaires
A. Nouvelle classification
B. Nouvelles directives
i. Développements et données cliniques
ii. Démarches renforcées
a. Documents qualités et techniques
b. Le chargé d’affaire réglementaire
iii. Amélioration du suivi
a. Traçabilité accrue
b. Surveillance après commercialisation
c. Eudamed
IV. Enjeux pour les entreprises
A. Développement du marché
i. Marquage CE
ii. Accès au marché
iii. Obtention de la certification
B. L’adaptation des entreprises aux nouvelles directives
i. Gestion de sous-traitants, fournisseurs, collaborateurs
ii. Changements dans l’organisation des entreprises
V. Discussion
VI. Conclusion
VII. Bibliographie
Télécharger le rapport complet