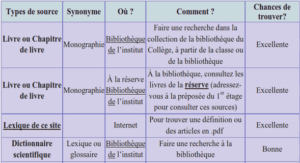
Télécharger le fichier pdf d’un mémoire de fin d’études
Diagramme configurationnel
La transition de spin de l’état bas spin (BS) à l’état haut spin (HS), implique une augmentation de la distance Fer-ligands. Il s’agit d’une occupation des orbitales anti-liantes eg par deux des six électrons de la couche d dans l’état HS, provoquant une répulsion des ligands et ainsi une augmentation de volume de la molécule. Dans le cas d’un octaèdre FeN6, les longueurs des liaisons Fer-ligands valent approximativement rBS = 1.95−2.00 Å dans l’état bas spin et rHS = 2.12−2.18 Å dans l’état haut spin. Les deux états moléculaires peuvent être représentés dans un diagramme configurationnel, comme illustré sur la figure I.4, par deux puits de potentiels adiabatiques décalés d’une distance ΔrHB ≈ 0.2 Å, correspondant à la différence de distance Fer-ligands entre les deux états de spin [67]. De plus, les concavités des deux puits sont différentes, ce qui est principalement attribué à un changement des fréquences de vibration de l’octaèdre lors de la transition de spin. Selon l’axe d’énergie E, les minima d’énergies des deux puits sont décalés d’une valeur ΔEHB0 = EHS0 − EBS0, où Ei0 (i=HS ou BS) est l’énergie du niveau de point zéro, égale à la somme des énergies électronique et vibrationnelle à température nulle : E0 = Eel + Evib(T = 0K). (I.1)
Sauf quelques exceptions [88, 89, 90], l’énergie du point zéro de l’état BS est in-férieure à celle de l’état HS, l’état fondamental est alors l’état BS à T = 0 K. Pour passer de l’état BS à l’état HS, il est nécessaire que la barrière d’énergie ΔEHB0 soit du même ordre de grandeur qu’un stimulus externe de l’ordre de kBT . A température non nulle, le potentiel thermodynamique à minimiser est l’enthalpie libre Gi = Hi − T Si que nous allons détailler dans la section suivante.
Thermodynamique de la transition de spin
On considère tout d’abord le cas d’un système dilué, i.e. d’un système constitué d’un grand nombre de molécules qui interagissent faiblement entre elles 1. Ce système est en contact avec un bain thermique et un barostat correspondant à l’ensemble de Gibbs (T , P , N). Le potentiel thermodynamique pertinent à minimiser est alors l’enthalpie libre. La variation de cette quantité peut s’écrire sous la forme : ΔG = GHS − GBS = ΔH − TΔS; (I.2)
où H est l’enthalpie, T la température, S l’entropie du système, ΔS = SHS − SBS et ΔH = HHS − HBS. La température d’équilibre entre les deux phases est déterminée lorsqu’il y a autant de molécules BS que de molécules HS, c’est-à-dire pour ΔG = 0 : Teq = ΔH (I.3)
1. On négligera donc dans un premier temps le terme qui prend en compte les interactions inter-moléculaires dans l’approche thermodynamique. Il faut cependant garder à l’esprit qu’en réalité, pour que le système rejoigne l’équilibre thermodynamique, il faut que les entités élémentaires qui consti-tuent le système microscopique échangent de l’énergie. Ces problèmes conceptuels sont récurrents et bien connus dans les modèles idéalisés (gaz parfait, oscillateurs harmoniques indépendants etc…).
Grâce à l’équation précédente, nous pouvons discuter de la stabilité des phases des systèmes dilués à transition de spin :
(1) Quand la température est inférieure à la température d’équilibre des deux phases, T < Teq, donc ΔH > T ΔS, le terme enthalpique est dominant et l’état le plus stable est l’état BS (GBS < GHS).
(2) Quand la température est supérieure à la température d’équilibre des deux phases, T > Teq, donc ΔH < T ΔS, le terme entropique est dominant et l’état le plus stable est l’état HS (GHS < GBS).
(3) Quand la température est égale à la température d’équilibre des deux phases, T = Teq, les termes enthalpique et entropique sont égaux (ΔH = T ΔS) et les deux phases sont présentes dans le système en même proportion.
D’après cette approche, il est clair que l’entropie peut être vue comme le moteur de la transition de spin thermo-induite en favorisant l’état HS. Pour donner une meilleure compréhension de la stabilité des phases des matériaux à transition de spin, il est indis-pensable de détailler les contributions contenues dans chaque terme apparaissant dans l’expression de la température d’équilibre Teq. Le terme enthalpique est généralement la somme des contributions électroniques et vibrationnelles [91], et peut s’écrire de la manière suivante : ΔH = ΔHvib + ΔHel ; (I.4)
La contribution électronique à l’enthalpie est souvent dominante, donc ΔH ≈ ΔHel.
De même, l’entropie se décompose en plusieurs termes de la manière suivante : ΔS = ΔSel + ΔSvib + ΔStrans + ΔSrot + ΔSconf ; (I.5)
L’entropie de translation ΔStrans, et de rotation ΔSrot, sont négligeables dans la plupart des composés à transition de spin, car généralement les molécules se trouvent dans un milieu solide, ce qui empêche leurs déplacements par translation ou rotation. De même, l’entropie de configuration ΔSconf est souvent négligeable. Cependant, une exception peut être faite dans le cas du composé Fe(pyrazine)[Pt(CN)4], où les ligands tournent autour d’un axe de rotation avec une vitesse dépendante de l’état de spin. Dans ce cas, la différence d’entropie de rotation n’est plus négligeable [109]. De plus, il a été reporté dans le composé [Fe(DAPP)(abpt)](ClO4)2 que l’entropie configurationnelle n’est pas négligeable, en raison de la présence de désordres induits par les ligands DAPP et l’ion ClO−4, contribuant alors légèrement à la différence d’entropie totale du système [108]. Concernant l’entropie liée à la contribution électronique ΔSel, ce terme lui même se décompose en deux parties : l’une est associée à la différence de dégénérescence orbitalaire ΔSorb, et l’autre à la différence de dégénérescence de spin ΔSspin. Les deux s’écrivent de la manière suivante :
ΔSorb = R ln 2LHS + 1 ; (I.6)
ΔSspin = R ln 2SHS + 1 ; (I.7)
où R = NAkB est la constante des gaz parfaits, kB la constante de Boltzmann, NA le nombre d’Avogadro, et L le moment orbital angulaire. Pour donner un ordre de grandeur à chacun de ces deux termes, prenons l’exemple de l’ion Fe(II) plongé dans un champ de ligands à symétrie octaédrique. Nous rappelons que l’état BS est un état diamagnétique possédant un spin électronique total S=0, alors que l’état paramagnétique (HS) ayant un spin S=2. Il en découle une différence d’entropie ΔSspin = R ln(5) = 13.38 J.mol−1.K−1. Souvent la symétrie autour de l’ion Fe(II) est plus basse et la dégénérescence orbitale est levée. Il en résulte que ΔSel ≈ ΔSspin > 0. L’entropie d’origine électronique favorise alors l’état HS et reste invariante en fonction de la température.
D’autre part, les vibrations de la sphère de coordination jouent un rôle central dans le mécanisme de la transition de spin. La différence d’entropie d’origine vibrationnelle ΔSvib dépend fortement de la différence des modes de vibration au sein de l’octaèdre entre les deux états de spin. L’entropie vibrationnelle s’écrit de la manière suivante : Svib(T ) = R− ln[1 − e−hνλ/kBT ] + kBT ehνλ/kBT ! ; (I.8)
La somme est effectuée sur tous les modes de vibration. Dans l’approximation des hautes températures ou des basses fréquences, hν << kBT , l’équation précédente se réduit à la forme suivante : X (I.9) Svib(T ) = R − ln[hνλ/kBT ] ;
La différence d’entropie vibrationnelle entre les deux états de spin devient alors : HSBS X νλBS ΔSvib = Svib − Svib = R ln ! ; (I.10)
Molécules dans un solide massif : Notion de Coopérati-vité
Nous avons évoqué, jusqu’à présent, le cas des molécules isolés ainsi que des sys-tèmes dilués. En milieu solide, les molécules interagissent entre elles à travers des interactions intermoléculaires. Il est important de noter que le changement drastique du volume et de forme quand une molécule passe de l’état BS à l’état HS, provoque une importante différence de volume dans le solide, ce qui conduit à une forte interac-tion élastique à long-portée influençant la totalité du solide. Deux types d’interactions élastiques sont possibles : soit par une contrainte en tension résultante d’une défor-mation provoquée par la commutation d’une molécule de l’état BS à l’état HS, soit une contrainte de compression lorsqu’une molécule commute vers l’état BS dans un environnement de molécules HS (voir Fig.I.6 a) et b)). Dans les deux cas, la com-mutation d’une molécule n’aura pas une répercussion uniquement sur les molécules se trouvant dans l’environnement immédiat de cette déformation locale, mais aussi sur l’ensemble du solide. Ces mécanismes élastiques sont à l’origine d’effets collectifs du réseau, pouvant provoquer une transition de spin abrupte (transition dite du pre-mier ordre) dans le cas où les interactions intermoléculaires sont suffisamment fortes (il conviendra de déterminer par rapport à quelle énergie caractéristique, les énergies liées aux interactions intermoléculaires doivent être plus importantes).
Le changement de volume d’une molécule induit un changement local des pro-priétés élastiques qui va se répercuter sur l’ensemble de réseau. Par analogie avec l’électrostatique, nous pouvons associer à ces effets locales une grandeur physique per-tinente appelée dipôle élastique, qui va générer un champ élastique dans tout l’espace matériel 2. Cette description des interactions élastiques simplifie la compréhension et l’interprétation des couplages élastiques en transformant un formalisme tensoriel en interactions dipôles-dipôles (voir Fig.I.6 c) et d)). Une interaction élastique entre N dipôles contribue largement à la coopérativité du réseau et donc aux commutations collectives au sein du solide. En effet, la propagation de contrainte élastique provoquée par une ou plusieurs molécules donne lieu à une réponse globale du réseau, appelé « pression image » [92]. La pression image est un ensemble d’interactions complexes et indissociables à courte et à longue portées [93].
Les différentes transitions de spin
Jusque dans les années 60, les transitions de spin étaient souvent des conversions graduelles de spin (voir Fig.I.7a) [30, 96]. Ce type de « transition » est très bien simulé avec le modèle des solutions régulières, présenté à la section I.2.3, que ce soit dans le cas des solides dilués ou des liquides. Chaque molécule est alors isolée et commute d’un état à un autre indépendamment de ses molécules voisines.
En 1964, Baker et Bobonich reportèrent un comportement magnétique « inhabituel » dans le composé [Fe(phen)2(NCS)2] à l’état solide [23], qui dévie d’une loi de type Boltzmann de peuplement des états. Ils conclurent que le changement d’état de spin observé dans ce composé était une transition du premier ordre impliquant l’émission ou l’absorption d’une chaleur latente (voir Fig.I.7b). Trois ans plus tard, König et Madeja réalisèrent une étude indépendante dans laquelle la transition du premier ordre fut confirmée pour le même composé[51]. La transition de spin abrupte fut ensuite attribuée à des effets coopératifs d’origine élastique.
Dans les systèmes dits très coopératifs, la transition de phase abrupte entre les deux états de spin, s’accompagne cette fois-ci, d’un cycle d’hystérèse, avec des températures de transition différentes en modes d’échauffement (T1+/2) et de refroidissement (T1−/2) (voir Fig.I.7c). En 1976, König et Ritter observèrent pour la première fois la présence d’un cycle d’hystérèse pour le composé [Fe(4,7-(CH3)2-phen)2(NCS)2] [53]. Ce phéno-mène, dû principalement aux fortes interactions intermoléculaires, est directement lié à l’existence d’états métastables dans lesquels le système peut rester piégé.
En 1981, l’existence d’une transition de spin en deux étapes fut observée par Ze-lentsov, pour un complexe de Fe(III) de 2-bromo-salicylaldehyde-thiosemicarbazone (voir Fig.I.7d) [98, 99]. Dans le cas des complexes polynucléaires [100], Sasaki et Kam-bara attribuèrent la transition de spin en deux étapes à des interactions antagonistes entre les distorsions élastiques existants au sein du composé et les contraintes élas-tiques qui se propagent dans tout le réseau. Cependant, il existe d’autres origines à ce phénomène. En effet, la présence de plusieurs sites non équivalents, c’est-à-dire l’exis-tence de centre métallique plongé dans des champs de ligands possédant des symétries différentes, mène à favoriser la transition de spin d’une partie de ces cations à une tem-pérature différente à celle des autres, d’où la transition de spin en plusieurs étapes. D’un point de vue théorique, il a également été montré qu’il pouvait s’agir d’une compétition entre les interactions élastiques à courte et à long-portée [101, 102, 103]. Finalement, nous mentionnons la présence de transition de spin incomplète [24], où une fraction résiduelle de molécules HS (resp. BS) existe à basses températures (resp. à hautes températures) (voir Fig.I.7e). Ce type de transition que nous détaillons par la suite, est observé à l’échelle nanométrique, mais également dans les matériaux mas-sifs. Ce phénomène peut avoir diverses origines, parmi lesquelles un effet de trempe thermique peut empêcher de rejoindre l’état BS, un désordre structural ou un effet de surface peut « geler » partiellement l’état de spin à basse température.
Approche macroscopique et mésoscopique
Les modèles thermodynamiques
Dans l’approche thermodynamique présentée précédemment (dans la section I.2.3), les systèmes dilués présentaient tous des conversions de spin graduelles. Cependant, à l’état solide, les interactions intermoléculaires jouent un rôle important dans la com-mutation moléculaire et leur considération théorique est indispensable pour simuler le cas des transitions de phase du premier ordre. En 1972, Slitcher et Drackamer intro-duisirent un terme phénoménologique non linéaire ( nHS(1 − nHS)) dans l’enthalpie de Gibbs (Eq.I.12) pour prendre en compte de manière effective et donner une des-cription à l’échelle macroscopique des interactions intermoléculaires [62]. En effet, par analogie avec le magnétisme, on peut faire apparaitre un tel terme en réalisant un développement de Taylor en nHS (paramètre d’ordre) jusqu’à l’ordre 2 : G = α + βnHS + γn2HS − T Smel = α + (β + γ)nHS − γnHS(1 − nHS) − T Smel (I.16)
= GBS + (GHS − GBS)nHS + nHS(1 − nHS) − T Smel = nHSGHS + (1 − nHS)GBS + nHS(1 − nHS) − T Smel ; avec est un paramètre phénoménologique associé à la coopérativité du réseau. La recherche des minima de l’enthalpie libre de Gibbs, donnée par ∂G T,P = 0, nous ∂nHS permet de remonter à la relation T = f(nHS) : R ln nHS + ΔS 1−nHS T = ΔH + (1 − 2nHS) ; (I.17)
Il découle de cette équation une infinité des solutions dépendantes de à une tempé-rature de transition Teq(nHS = 0.5) = ΔH/ΔS. Néanmoins, nous pouvons discuter de la stabilité des positions d’équilibre de l’enthalpie libre G(nHS) au travers de l’étude du signe de la dérivée seconde de l’enthalpie libre : ∂2G ! = −2 + 4RTeq ; (I.18)
D’après cette équation, nous pouvons dégager trois situations représentées sur la figure I.8 :
(1) Cas d’une faible interaction intermoléculaire lorsque < 2RTeq, alors ∂2G T,P > ∂nHS2 0. Le point se trouve sur un minimum de l’enthalpie libre de Gibbs. La transition est ainsi graduelle (conversion de spin) sans la présence d’un cycle d’hystérésis.
(2) Cas d’une forte interaction intermoléculaire lorsque > 2RTeq, alors ∂2G T,P < ∂nHS2 0. Le point se trouve sur un maximum de l’enthalpie libre de Gibbs. La transition est ainsi abrupte avec la présence d’un cycle d’hystérésis. Il est important de noter que la température de transition en mode chauffage (T +) ne correspond pas à celle en mode refroidissement (T −). Ceci est due à la présence des états métastables représentés sur la figure I.9.
(3) Cas intermédiaire lorsque = 2RTeq, alors ∂2G T,P = 0, il s’agit d’un point ∂nHS2 d’inflexion de la courbe de transition. Dans ce cas, la transition est abrupte sans l’apparition d’un cycle d’hystérésis.
Effet de la réduction de la taille : présentation générale
Ces dernières décennies, la question des effets de la réduction de taille sur les propriétés de la matière condensée devient de plus en plus récurrente dans les com-munautés scientifiques, en particulier après l’élaboration et la synthèse de différents objets de taille nanométrique. C’est le changement des propriétés physico-chimiques des nanoobjets par rapport aux matériaux massifs, constaté dans les différentes me-sures expérimentales, qui a poussé les chercheurs à s’engager dans l’étude des effets de taille, plus particulièrement, des effets et des caractéristiques spécifiques 7 des sur-faces et interfaces. En effet, les nanomatériaux possèdent en générale des structures électroniques et vibrationnelles, ainsi qu’une morphologie différente du matériau massif correspondant conduisant à l’émergence de nouvelles propriétés optiques, magnétiques, mécaniques intéressantes et à la modification des mécanismes de transport électrique et phononique. La réduction de la taille peut également provoquer l’altération de certaines caractéristiques initialement présentes dans le matériau massif ou la modification ou la disparition non désirée de phénomènes critiques telles que les transitions de phase. D’un point de vue théorique, la prédominance des effets de surface avec la réduction de la taille au détriment des propriétés dites de volume vient de la modification du rapport du nombre d’atomes situés en surface par rapport au nombre d’atomes situés en volume. En effet, à grande taille, ce rapport est négligeable, car indépendamment de la forme du matériau, le nombre des atomes volumiques est plus important que les atomes surfaciques, donc les atomes de surface peuvent être généralement négligés par rapport aux atomes de volume. En d’autres termes, les propriétés spécifiques de la surface sont négligeables vis-à-vis des propriétés volumiques. Ce n’est plus le cas lorsque l’une des dimensions du système devient trop « petite » devant une longueur ca-ractéristique associée aux propriétés ou au phénomène de transport considérés (libre parcours moyen des phonons, des électrons, longueurs de corrélation spatiales d’ob-servables associées à une transition de phase, etc …). Les entités situées à la surface ne peuvent alors plus être négligées. On peut alors parler d’un effet de taille car les propriétés physico-chimiques des matériaux changent sous la dominance des effets spé-cifiques de surface/interface ou/et des effets de confinement (dus à la diminution de la quantité de matière ou à la limitation dans l’espace du transport d’une certaine quantité physique).
Le but de cette section est d’exposer très brièvement les effets de la présence de surfaces et de confinement spatial dans les propriétés physico-chimiques des nanomaté-riaux, afin de prédire dans un premier temps les conséquences possibles de la réduction de taille sur la thermodynamique des matériaux à transition de spin.
Effets de surfaces
Influence sur les spectres vibrationnels des nanomatériaux
Au début de XXe siècle, Born et von Karman décrivirent les vibrations atomiques et développèrent ce que l’on appelle la théorie dynamique des cristaux [132]. Dans un premier temps, les effets de surface sur la dynamique de réseau furent négligés. Cependant, l’amplitude des oscillations des atomes de surface est plus importante que celle des atomes de cœur du fait de la présence de liaisons pendantes. Cela implique la possible existence de modes vibrationnels spécifiques de la surface dans le spectre de vibration du volume. Parmi ces modes, les modes de Rayleigh sont les plus connus car ils sont largement étudiés en géophysique dans le domaine de l’analyse des dégâts causés par certaine catégorie de séismes. Ces modes localisés en surface qui s’atténuent exponentiellement dans le volume subsistent également à une échelle nanométrique, leur considération est cruciale pour déterminer les propriétés vibrationnelles des na-nomatériaux. Afin d’exposer la contribution de surface dans les propriétés vibrationnelles, consi-dérons le cas de vibration d’un cristal monoatomique cfc avec un potentiel de Lennard-Jones. Pour ce cristal, la structure de bandes de phonons a été calculée et représentée dans la zone de Brillouin de surface de type (111) [133] (voir Fig.I.14). Cette relation de dispersion de phonons montre l’implication des modes de surface (111) notés S dans le spectre vibrationnel du volume. L’existence des modes vibrationnels de surface près des modes du volume (les zones sombres) conduit certainement à une dominance des effets de surface dans les propriétés vibrationnelles des nanocristaux, car à une échelle nanométrique, les modes du volume se raréfient au détriment des modes surfaciques. Les modes de surface existent même si la structure et les constantes de forces des atomes de surface sont identiques à ceux des atomes de volume. En absence de surface ou dans les matériaux massifs, les modes Si de surface disparaissent (en moyenne), il n’y a donc que les modes de volume qui déterminent les propriétés vibrationnelles du matériau.
Influence sur la densité électronique des nanomatériaux
La présence de surface peut également influencer la densité électronique des élec-trons délocalisés dans les nano-objets conducteurs ou semi-conducteur. L’étude des effets électroniques de surface est le point-clé pour comprendre la plupart des phé-nomènes physico-chimiques de surface, tels que la réaction chimique, la croissance cristalline, les problèmes d’adhésion, etc…
La description de l’influence de surface sur la densité électronique des nanomaté-riaux nécessite de résoudre le problème des électrons libres dans une boîte délimitée par des surfaces dans toutes les directions (x, y, z). La fonction d’onde des électrons, solution de l’équation de Schrödinger, doit valider les conditions d’annulations en bor-dure de la boîte, une condition imposée par la création des surfaces. La fonction d’onde s’écrit alors de la manière suivante : →− s r niπ 8 sin kxx sin kyy sin kzz ; ki = ψ k (→−) = (I.46)
où les ni→−sont des entiers strictement positifs. V = Lx × Ly × Lz est le volume de la boîte et k = kxeˆx + kyeˆy + kzeˆz le vecteur d’onde dans l’espace réciproque.
Partant de la fonction d’onde, on peut remonter à la densité électronique n près de la surface en calculant tout d’abord la probabilité de présence d’un électron par le module carré de la fonction d’onde. Il en découle la densité électronique suivante : kF3 (sin x − x cos x) ! n(z) = 1 − 3 ; x = 2kF z (I.47)
où kF est le rayon de Fermi de la sphère occupée par les états électroniques (voir Fig.I.15). Les détails du calcul se trouvent dans la référence [135]. Loin de la surface, la densité électronique correspond exactement à celle du volume (n(z) = n∞). À l’approche de la surface, en revanche, cette densité oscille et ne correspond pas à la densité électronique du volume, car la présence de surface conduit à un excès de charges électroniques hors de la surface ainsi qu’un excès de charge positive près de la surface du matériau (voir Fig.I.15). Par conséquence, il y a création d’un dipôle de surface. Le travail de sortie d’un électron est alors le travail nécessaire pour vaincre l’interaction électron-dipôle de surface.
Dans une autre approche basée sur la méthode de densité locale, il a été montré que la présence de surface conduit à l’apparition de nouveaux états de surface d’énergie différents des états du volume. Ces états localisés en surface, peuvent exister dans la bande interdite de la densité électronique volumique, comme par exemple dans le cas du cristal de Si(111) [136].
Effets de confinement
L’effet de confinement est un phénomène généralement apparent lorsque la dimen-sion du matériau est comparable au libre parcours moyen de la particule quantique. Dans ce cas, le cristal cesse de se comporter comme un milieu continu, conduisant à une quantification du spectre en énergie (par exemple, les zones sombres continues représentant la structure de bande phononique de volume montrées sur la figure I.14 deviendraient discontinues).
Dans le cas des phénomènes de transport des déformations dans un solide cristallin isolant, le libre parcours moyen de phonons est décrit comme la longueur moyenne que la particule phononique peut effectuer avant d’entrer en collision avec une impureté (défaut dans le solide) ou simplement avec un autre phonon. Lorsque le libre parcours moyen devient du même ordre de grandeur que l’une des dimensions spatiales du nanomatériau, les collisions s’effectuent principalement au niveau des surfaces ou des interfaces et de moins en moins au cœur du nano-objet. On assiste à un changement de mécanisme de transport : le système passe d’un régime diffusif à un régime balistique. Sachant qu’un phonon est une quasi-particule qui transporte de la chaleur, un régime balistique empêche aux quasi-particules de « communiquer » par collision et l’équilibre thermodynamique (dans ce cas thermique) est difficilement atteignable.
Pour résumer, les effets de confinement sont liés à deux phénomènes : la diminution de la quantité de matière qui entraîne la discrétisation de la structure de bande (ici phononique) et une extension spatiale limitée du système dans certaines directions de l’espace qui a tendance à modifier les mécanismes de transport (et donc les comporte-ments collectifs). De plus, le confinement peut affecter la densité d’états vibrationnels en modifiant la position des modes de vibration du nano-objet. Par exemple, il a été observé un durcissement des modes acoustiques et un ramollissement des modes op-tiques en présence du confinement spatial [137, 138, 139, 140, 141, 142, 83]. Ce dernier est susceptible de rendre caduque la théorie de l’élasticité linéaire ou de l’approxima-tion continue (valable uniquement lorsque les longueurs d’onde associées aux modes de vibrations de basses fréqueneces sont grandes devant les paramètres de maille du cristal). Dans ce cas les grandeurs physiques qui caractérisent les propriétés élastiques des matériaux massifs (vitesse du son, module d’Young etc…) ne sont plus définies et ne sont plus pertinentes lorsque l’on souhaite analyser les propriétés mécaniques des nano-objets.
Conséquences possibles sur la transition de spin des na-nomatériaux
Les effets de surface et de confinement spatial peuvent alors influencer les propriétés vibrationnelles, électroniques, mais également magnétiques et optiques des nanoma-tériaux. La question pertinente à se poser est : pouvons-nous nous attendre à une influence de la surface et du confinement spatial sur la thermodynamique et la stabi-lité des phases des nanomatériaux à transition de spin ? En effet, Gibbs [174] définit les quantités de surface comme des grandeurs en excès par rapport aux propriétés vo-lumiques. Parmi ces grandeurs, deux ont une grande importance car elles permettent de définir le travail lié à la création de surface (énergie de surface) et le travail de déformation de surface (contrainte de surface). Ces deux quantités dépendent prin-cipalement de l’excès de la densité électronique et des propriétés vibrationnelles et élastiques en surface. Cependant, nous ne nous attendons pas à un excès d’électrons en surface dans les matériaux à transition de spin, car dans ce type de matériaux, les électrons sont fortement localisés, et l’approximation du gaz d’électrons presque libres ne s’appliquent donc pas dans ce cas. Toutefois, le nuage électronique des com-plexes à transition de spin peut être déformé du fait de la présence d’une surface. Des dipôles électriques peuvent alors être induits et apparaître au niveau de ces sur-faces. Une description théorique de ces effets prenant en compte les redistributions de charges serait envisageable. La conséquence (ou la cause) possible de ces distorsions du nuage électronique en surface est une modification des longueurs de liaison (distance métal-ligand, paramètre de maille …) à l’approche des surfaces. Dans ce type de maté-riaux isolants, si l’on ne s’intéresse pas au chemin réactionnel amenant à la structure de surface observée expérimentalement, il est alors possible d’observer indirectement les réorganisations électroniques de surface au travers de l’étude des changements des propriétés vibrationnelles. Même si les électrons restent fortement localisés en surface, les nouveaux modes de vibrations optiques et acoustiques générés à la fois par la re-distribution de charges et par la brisure de l’invariance par translation des propriétés physiques vont quant à eux se délocaliser dans le coeur du matériau de taille finie et seront susceptibles d’affecter le couplage électron-phonon, les propriétés élastiques et thermomécaniques.
Nanomatériaux à transition de spin : les consta-tations expérimentales
Initialement, l’une des motivations majeures ayant poussé la communauté scienti-fique à étudier les matériaux à transition de spin est la présence possible du phénomène de bistabilité dans la mesure de certaines grandeurs physiques. Cette propriété confère à ces matériaux un potentiel applicatif certain dans le domaine du stockage d’informa-tion [143]. Dans ce contexte, la communauté scientifique, en particulier Olivier Kahn, se sont efforcés de développer des dispositifs de stockage d’information reposant sur l’effet mémoire de ces matériaux [144]. Très rapidement, la nécessité de miniaturiser ces objets commutables pour leur intégration dans des dispositifs de taille nanométrique a été pointée du doigt au début des années 2000. Dès lors, la cours à la fabrication de nano-objet transition de spin était lancée : synthèse de nanoparticules de formes diverses, conception de films minces, nano-structuration … [145].
Dans cette partie, nous présentons une historique brève et succincte de l’élaboration de nano-objets à transition de spin, plus particulièrement de nanoparticules et de films minces. Nous nous attarderons sur l’évolution avec la diminution de la taille de leurs propriétés physiques. Ce sera l’occasion d’effectuer des comparaisons avec les effets de réduction de taille qui ont pu être observés dans d’autres systèmes présentant des transformations de phase.
Cas des nanoparticules à transition de spin
C’est en 2008 que des nanoparticules [Fe(NH2-trz)3)](Br)2 de la famille des Fe(II)-triazole, sont synthétisées à l’aide de la technique des micelles inverses [68]. Ces nano-particules sphériques de taille moyenne 69 nm montrent la présence d’un cycle d’hysté-résis thermique à la température ambiante. Ensuite, Coronado et al [69] démontrèrent que la technique de micelles inverses pouvait être appliquée aux polymères à transition de spin, tel que [Fe(Htrz)2(trz)](BF4), pour contrôler la croissance des cristallites. Les nanoparticules obtenues de taille moyenne 10 nm révèlent un cycle d’hystérèse de lar-geur 43 K à une température de transition supérieure à la température ambiante. En 2010, des nanoparticules de sels de Fe(II) triazole sont préparées par microémulsion [70], la taille moyenne obtenue est environ 6 nm, et un cycle d’hystérèse est de nouveau constaté dans ces nanoparticules mais d’une largeur inférieure à 30 K. De plus, la tran-sition est incomplète avec l’apparition de 33 % de fraction HS à basse température. L’étude en taille des [Fe(Htrz)2(trz)](BF4) s’est poursuivie en 2015 avec cette fois-ci des nanoparticules de taille allant de 16 à 4 nm [71], les auteurs ont observé une transition abrupte pour ces nanoparticules avec une diminution de la largeur du cycle d’hystérésis de 24 à 13 K en diminuant la taille, une large hystérèse est alors préservée à l’échelle nanométrique (voir figure I.17a). Cependant, la forme et la taille de ces objets ont été remises en question par Murgui et al, qui affirment dans une publication [72] que la méthode de synthèse utilisée ne permettrait d’obtenir que des nano-bâtonnets d’une longueur pouvant atteindre quelque dizaines de nanomètres. Par ailleurs, d’autres tra-vaux, tels que ceux de Durand et al [73], montrent la présence d’un cycle d’hystérèse pour des nanoparticules de 3-4 nm que l’on a fait croître dans des pores de silice. En parallèle, Létard et al [74, 75] observent une perte d’hystérèse et une transition graduelle pour des nanoparticules de [Fe(NH2-trz)3]Br2 en dessous d’une taille critique de 50 nm (voir figure I.17 (b)), tandis que des mesures d’absorption d’UV réalisées dans notre équipe montrent l’existence d’une transition abrupte avec la présence d’un cycle d’hystérèse dans des nanoparticules de 10 nm de [Fe(NH2trz)3](tos)2 [76] mais une absence de l’effet mémoire pour des particules de 3-4 nm [77, 11, 78].
Figure I.17 – a) Évolution du produit de la susceptibilité magnétique avec la tempé-rature (χT ) en fonction de la température pour différentes tailles de nano-particules. Pour le composé [Fe(pyrazine)Pt(CN)4], il est possible d’observer l’effet mémoire pour le matériau massif (bulk). En diminuant la taille, il y a une perte du cycle d’hystérésis, une baisse de la température de transition et l’apparition d’une fraction résiduelle HS à basse température (courbe violet : 14 nm ; courbe verte : 7 nm)[13]. b) Représentation de l’aimantation M en fonction de la température pour un ensemble de nanopar-ticules de 3-4 nm du composé [Fe(pyrazine)Ni(CN)4] dans une matrice de chitosan (bio-polymère) [81].
D’autre part, des effets de taille significatifs ont été mentionnés par Volatron et al [13] pour des nanoparticules de 7 et 14 nm, en remarquant une perte de l’effet mé-moire avec une importante fraction résiduelle HS à basse température accompagnée d’une chute drastique de la température de transition. Par ailleurs, Larionova et al [81] montrèrent une transition de spin très incomplète avec la présence significative de sites inactifs, ainsi que la présence d’un cycle d’hystérésis pour des nanoparticules [Fe(pyrazine)Ni(CN)4] de taille 4 nm dont la croissance s’est effectuée dans les pores d’une matrice biopolymérique (chitosan). La miniaturisation de particules du composé [Fe(pz)Pt(CN)4] a été poursuivie par Boldog et al [80], qui ont pu élaborer des nano-particules d’une dizaine de nanomètre. Les premiers signes d’un effet de taille sur ces composés ont été remarqués en constatant des propriétés magnétiques différentes de celles observées pour les systèmes massifs. Des observations différentes ont été faites par Peng et al. [14] sur des nanoparticules de la même famille (clathrates de Hoffmann) mais obtenues par des méthodes de micelles inverses. Un comportement non monotone de la température de transition est constaté avec la diminution de la taille ainsi que la réapparition d’un cycle d’hystérésis pour des nanoparticules de très petites tailles (< 2 nm).
Cas des films minces à transition de spin
La première préparation de films minces SCO concerne l’élaboration d’un assem-blage bidimensionnel en utilisant la technique de Langmuir-Blodgett (LB) [146]. Cette technique consiste à remplacer le phen de cis-Fe(phen)2(NCS)2 par X2Yphen contenant des chaînes hydrophobes pour créer, après compression, des monocouches de molécules SCO à l’interface eau/air [147, 148]. Plus tard, la croissance des films minces SCO a été réalisée sur des surfaces solides en utilisant l’approche d’assemblage séquentielle, le revêtement par centrifugation (spin coating en Anglais) , le « drop casting », ou la technique d’évaporation thermique.
En 2006, l’approche d’assemblage séquentielle [149, 150] a été utilisée pour la pre-mière fois par Cobo et al. [9] au cas des matériaux SCO. Cette technique permet de construire un multicouches 3D de réseaux de coordination en assemblant d’une manière séquentielle des couches bidimensionnelles (couche par couche) reliées entre elles par un ligand pontant dans la troisième dimension. Des films minces de la famille de cla-thrates de Hoffmann, en particulier le réseau de coordination [Fe(pyrazine)(Pt(CN)4)], ont été élaboré en utilisant l’approche d’assemblage séquentielle. Dans la même année, Kuroiwa et al. [151] reportèrent l’élaboration par la technique « drop casting » des films minces du réseau de coordination 1D basé sur le composé triazole Fe(II), qui possède une large hystérésis autour et au dessus de la température ambiante. Le revêtement par centrifugation « spin coating » est l’une des méthodes la plus utilisée pour faire croître des films minces à épaisseur contrôlée. D’abord, elle a été utilisée par Bous-seksou et al. [152] et appliquée au cas des matériaux SCO. Une faible coopérativité a été observée à une épaisseur de 30 nm du film mince [Fe(hptrz)3](OTs)2 (hptrz=4-heptyl-1,2,4-triazole et OTs=tosylate) revêtu par centrifugation sur verre et étudié par résonance plasmonique de surface (voir Fig.I.18 ) [153]. Finalement, la technique d’évaporation thermique permet d’obtenir des films minces d’épaisseur allant du nano-mètre jusqu’au micromètre. En utilisant cette méthode, Shalabaeva et al. [10] ont pu faire croître des films minces de hautes qualités du complexe [FeII(HB(tz)3)2](tz=1,2,4-triazol-1-yl) (voir Fig.I.19a). Ce travail a mis en évidence une augmentation de 3 K de la température de transition lorsque l’épaisseur de la couche mince passe de 200 nm à 40 nm (voir Fig.I.19b). Ce résultat est important car jusque maintenant on observait systématiquement une diminution de la température de transition avec la diminution de la taille. Ce comportement original 8 a été simulé numériquement à l’aide d’un modèle nanothermodynamique prenant en compte à la fois la différence d’énergie de surface entre les deux phases HS et BS et la forte anisotropie des propriétés élastiques et mécaniques lorsque le système change d’état de spin. Toutefois, si l’explication du mécanisme conduisant à une inversion du changement de la stabilité des phases est très certainement pertinente, la simulation avec le modèle nanothermodynamique n’est pas pleinement satisfaisante car la prise en compte des effets de surface/interface se fait au moyen d’un paramètre phénoménologique qui ne distinguent pas la contribution du défaut de coordination (liaison pendantes) et celle des relaxation de surface/interface.
Comparaison avec d’autres matériaux
La réduction de la taille des nanoobjets à transition de spin favorise la stabilité d’une phase (HS ou BS) et donc change la température de transition. La modification des propriétés de commutation, en particulier, le changement de stabilité des phases semblent être dépendants de la nature matériaux à transition de spin (complexes mo-léculaires, réseaux et polymères de coordination), de la forme et de la mise en forme du nano-objets ainsi que de l’environnement extérieur [154]. Ces observations sont com-parables à celles effectuées sur d’autres matériaux présentant un effet de taille, tels que les oxydes métalliques, ou les films semi-conducteur de la famille des hydrocarbures aromatiques polycycliques tels que le pentacène. En effet, l’oxyde métallique binaire Al2O3 subit une transformation de phase structurale αAl2O3 → γAl2O3 en dessus d’une certaine surface spécifique (125 m2.g−1) en raison d’une énergie de surface qui favorise la phase γAl2O3 à l’échelle nanométrique (voir Fig.I.20a) [155, 156]. De plus, en dessous d’une taille de particule de 14 nm, la forme anatase du TiO2 est énergéti-quement plus stable que sa forme rutile en raison d’une énergie de surface inférieure pour l’anatase (γA < γR) [157]. En ce qui concerne la transformation structurale de la phase triclinique à la phase orthorhombique avec la diminution de l’épaisseur ob-servée dans les films minces de pentacène [158], un modèle thermodynamique révèle que l’énergie libre de surface et l’énergie élastique provenant de la pression intérieure induisent une cristallisation de la phase orthorhombique plutôt que celle triclinique à l’échelle nanométrique (voir Fig.I.20b) [159]. Il est maintenant question de savoir pour-quoi la phase HS (resp. BS) est favorisée la plupart du temps dans les nanoparticules (resp. dans certains films minces) induisant une diminution (resp. augmentation) de la température de transition. Une étude théorique utilisant des approches issues de la physique statistique ou des modèles atomistiques est envisageable afin de clarifier le rôle des quantités de surface/interface et de confinement spatial dans la stabilité de phases des nanomatériaux à transition de spin.
|
Table des matières
I Introduction
I.1 Historique
I.2 Théorie de champ cristallin et de champ de ligands
I.2.1 Exemple du Fe(II)
I.2.2 Diagramme configurationnel
I.2.3 Thermodynamique de la transition de spin
I.2.4 Molécules dans un solide massif : Notion de Coopérativité
I.2.5 Les différentes transitions de spin
I.3 Approche macroscopique et mésoscopique
I.3.1 Les modèles thermodynamiques
I.3.2 Modèles élastiques
I.3.3 Approche microscopique de la transition de spin : les modèles de type d’Ising
I.3.4 Modèles microscopiques élastiques
I.4 Effet de la réduction de la taille : présentation générale
I.4.1 Effets de surfaces
I.4.2 Effets de confinement
I.4.3 Conséquences possibles sur la transition de spin des nanomatériaux
I.5 Nanomatériaux à transition de spin : les constatations expérimentales
I.5.1 Cas des nanoparticules à transition de spin
I.5.2 Cas des films minces à transition de spin
I.5.3 Comparaison avec d’autres matériaux
I.6 Modélisation des effets de la réduction de taille dans les matériaux à transition de spin
I.6.1 Modélisation des effets de surface solide-vide
I.6.2 Modélisation des effets d’interface solide-solide
I.6.3 Modélisation de l’influence des effets de surface et d’interface sur les courbes de transition de spin
I.7 Dynamique de réseau des nanomatériaux à transition de spin
I.7.1 Effets de taille sur la dynamique de réseau : Études expérimentales
I.7.2 Modélisation de la dynamique de réseau
I.8 Problématiques et objectifs de thèse
II Étude numérique de la dynamique de réseau des nanomatériaux à transition de spin
II.1 Introduction
II.2 Méthode numérique (DM) pour calculer la (vDOS)
II.2.1 La fonction d’autocorrélation des vitesses et la densité d’états vibrationnels
II.2.2 Intégration de positions et vitesses par la méthode de Verlet
II.2.3 Thermostat-barostat de Nosé-Hoover
II.2.4 Extraits de propriétés thermoélastiques
II.3 Application à une structure cubique à motifs octaédrique
II.3.1 Effet du potentiel angulaire sur l’élasticité du système
II.3.2 Modification du champ de forces : choix du potentiel intermolé- culaire
II.3.3 Propriétés vibrationnelles du matériau massif
II.3.4 Densité d’états vibrationnels dans le cas des films minces
II.3.5 Étude en taille des quantités thermodynamiques
II.3.6 Densité d’états vibrationnels dans le cas de nanoparticules
II.4 Étude de la dynamique de réseau des nanomatériaux à transition de spin par des méthodes de matrice dynamique
II.4.1 Application à une structure octaédrique bidimensionnelle
II.4.2 Étude des effets de taille par la matrice dynamique
II.5 Conclusions
III Étude quantitative des énergies et contraintes de surface dans les nanomatériaux SCO
III.1 Introduction
III.2 Approches théoriques des surfaces et des interfaces
III.2.1 Aspects thermodynamiques et stabilité des phases
III.2.2 Estimation de ∆S et ∆H du système massif
III.2.3 Énergie de surface
III.2.4 Élasticité d’interface
III.3 Énergie de surface : Estimation d’ordre de grandeur
III.4 Application à la structure cubique à motifs octaédriques
III.4.1 Calculs des propriétés mécano-thermodynamiques du matériau massif
III.4.2 Modélisation des énergies de surfaces (100)
III.4.3 Calcul des contraintes d’interface HS/BS dans le cas d’un faible désaccord de paramètre de maille
III.5 Conclusions
Conclusion générale
A Estimation de l’incertitude due aux artefacts numériques
B Calcul de l’énergie de surface par la méthode des liaisons coupées
C Films minces dans le cas de grands désaccords élastiques
Bibliographie
Télécharger le rapport complet