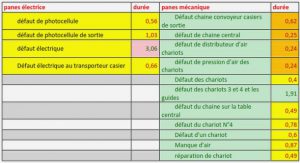
Le choix du renfort et de la matrice
Le renfort confère au matériau composite ses caractéristiques mécaniques telles que sa rigidité, sa résistance en rupture ou encore sa ténacité. Pour certaines applications industrielles, il peut être choisi pour ses propriétés thermiques ou électriques. Il existe plusieurs familles de fibres utilisées comme renfort :
➢ Fibre de carbone
➢ Fibres de verre
➢ Fibres d’aramides (ex. Kevlar)
➢ Fibres céramiques (ex. Bore, SiC, SicTi)
Dans le cadre de la thèse, seul le renfort à fibre de carbone est décrit.
Le renfort : fibres de carbone Largement utilisées dans le secteur aérospatial en raison de ses hautes performances mécaniques, les fibres de carbone sont obtenues à partir de polycrylonitrile (PAN) ou de brais mésophases [Dupupet2008]. Cependant, il est industriellement plus intéressant d’élaborer la fibre de carbone à partir du précurseur PAN en raison d’une meilleure maîtrise du procédé de fabrication. Suivant les critères de performance choisis pour la fibre (ex. Haute résistance à la rupture (HR) – Haut module d’élasticité (HM)), la fibre de carbone peut être produite suivant deux méthodes [Figure 1.2]. Le précurseur PAN, disponible sous la forme d’un monofilament obtenu par filage et étirage, subit une oxydation sous ambiance air dans un four à une température comprise entre 200◦C et 300◦C. Le précurseur change physiquement et chimiquement pour atteindre un état infusible thermiquement stable. Une étape de carbonisation sous azote à une température comprise entre 1200◦C et 1500◦C permet d’obtenir un filament orienté axialement en raison du réarrangement de sa microstructure. De plus, le sous-produit perd environ 50% de sa masse. À l’issue de cette étape, les fibres classées (HR) ont une teneur en carbone comprise entre 90% et 97%. Dans le cas où l’on souhaiterait produire des fibres à haut module d’élasticité (HM), une phase de graphitisation est nécessaire. Cela consiste en un second traitement thermique entre 2000◦C et 3000◦C afin d’éliminer les atomes d’azote et d’oxygène. La teneur en carbone obtenue dépasse les 99%. Finalement, les filaments subissent un traitement de surface pour améliorer leur adhérence à la matrice et une phase d’ensimage pour augmenter la résistance au frottement lors des étapes de mise en oeuvre textile (filage des mèches, tissage du renfort). Les fibres de carbone sont classées suivant leurs propriétés mécaniques en traction : HM pour haut module d’élasticité, IM pour module d’élasticité intermédiaire et HR pour haute résistance à la rupture. Une comparaison des propriétés mécaniques en traction des trois classes de fibres de carbone est proposée en [Figure 1.3]. Les fibres sont disponibles en plusieurs tailles : les fibres courtes (3, 0 à 4, 5 mm), les fibres longues (de longueur généralement supérieure à 6, 0 mm) et les fibres continues (CLFT) dont la longueur est de l’ordre de grandeur de la dimension d’une pièce composite afin de préserver la continuité du renfort [Lucas+2007b]. Dans le cadre de la thèse, on s’intéressera uniquement aux fibres continues et longues ainsi qu’aux procédés associés. Les filaments de renfort sont rassemblés pour former un fil ou une mèche. La structure de la mèche varie suivant les types de fibres utilisés, mais aussi du degré de mélange de ces fibres au sein d’une mèche. Cette dernière peut s’intégrer dans une structure constituant un « roving » [Figure 1.4].
Les matrices thermoplastiques Les matrices organiques employées dans la mise en œuvre des pièces composites (CMO) jouent le rôle de liant afin de conserver la disposition géométrique des fibres tout en transmettant les sollicitations auxquelles la pièce est soumise. Les résines doivent être suffisamment déformables, compatibles avec les fibres et avoir une densité relativement faible pour que le matériau conserve des caractéristiques mécaniques spécifiques élevées. Généralement, on considère deux familles de polymères : les thermodurcissables (TD) et les thermoplastiques (TP). Les matrices thermodurcissables sont majoritairement employées (à hauteur de 70%) dans la mise en œuvre de pièces composites même si ce niveau décroît graduellement au profit des matrices thermoplastiques. En cause notamment la réglementation « REACH » qui impose aux industriels un contrôle de plus en plus drastique dans l’utilisation des monomères additifs et autres molécules intervenant dans la synthèse des polymères. Les résines à base phénolique, qui rentrent dans certaines compositions de matrices thermodurcissables, vont être prochainement interdites d’utilisation en raison des risques avérés pour la santé. L’utilisation des polymères thermoplastiques en tant que matrice de substitution dans la fabrication de pièces composites constitue donc une alternative intéressante. Un polymère thermoplastique est caractérisé par de longues chaînes linéaires de monomères faiblement ramifiées chimiquement séparées entre elles. Ces chaînes sont liées entre elles par des liaisons faibles de type Van der Waals, dipôle-dipôle ou hydrogène. Ainsi, du fait de la nature de ces liaisons, la structure enchevêtrée du polymère ne résulte pas d’une réaction de polymérisation, mais d’un phénomène de repliement des chaînes lors du chauffage du thermoplastique. Lors du refroidissement, les chaînes se rigidifient et solidifient le matériau. Contrairement aux thermodurcissables, dont la polymérisation induit la formation d’un réseau réticulé par l’établissement de liaisons covalentes, les thermoplastiques peuvent subir plusieurs cycles de chauffage/refroidissement. La synthèse du polymère thermoplastique peut être obtenue suivant plusieurs voies de polymérisation : réaction de polyaddition, copolymérisation ou polycondensation. Les mécanismes d’homopolymérisation (enchaînement d’un même monomère) et de copolymérisation (polymérisation de deux monomères) interviennent majoritairement dans l’établissement d’une chaîne de monomères [Paris2011]. Un thermoplastique peut se présenter suivant deux structures : amorphe et semicristalline [Waddon+1987]. Sa mise en œuvre s’effectue à l’état caoutchoutique ou visqueux et se décompose en trois étapes :
➢ Le chauffage du polymère pour augmenter la mobilité des macromolécules. La température doit être supérieure à la température de transition vitreuse (Tg) pour les thermoplastiques amorphes et supérieure à la température de fusion (Tm) pour les thermoplastiques semi-cristallins.
➢ Le formage du polymère à l’état fondu.
➢ Le refroidissement du polymère qui doit être maîtrisé dans le cas d’une structure semi-cristalline pour permettre une cristallisation optimale et l’obtention de ses propriétés mécaniques et physico-chimiques.
Il existe dans le commerce trois catégories de thermoplastiques : les polymères de grande diffusion (PP), les polymères techniques (PC,PET,PBT…) et les polymères à hautes performances utilisés principalement dans les secteurs de pointe tels que l’aéronautique. Dans cette dernière catégorie, on trouve notamment le PEEK, le PEI et le PPS. Ils présentent plusieurs avantages par rapport aux thermodurcissables [Bathias+2009 ; Berthelot2012 ; Bessard2012] :
➢ Une tenue à l’impact et aux chocs supérieure.
➢ Une meilleure résistance chimique.
➢ Une réutilisation possible sous certaines conditions. La synthèse de ces polymères n’engendre pas un changement d’état permanent (du fait des réactions de polyaddition). Ils peuvent donc être à nouveau fondus et recristallisés à travers des cycles chauffage/refroidissement successifs.
➢ Une durée de stockage illimitée.
➢ Utilisable pour la mise en œuvre de pièces composites à géométries complexes.
➢ Des cycles de mise en oeuvre rapides
➢ Faible coût de production en règle générale à l’exception des thermoplastiques hautes performances tels que le PEEK (70 – 80 euros/kg).
Cependant, la température de mise en œuvre des polymères thermoplastiques est bien supérieure à celle requise pour les thermodurcissables. La viscosité à la température de mise en œuvre d’un thermoplastique est aussi plus élevée (100 à 1000 P a.s) que celle d’un thermodurcissable (0,1 à 10 P a.s) [Figure 1.5]. Cela engendre des difficultés dans la mise en œuvre des pièces composites. Finalement, le choix de la matrice pour la fabrication de pièces composites doit impérativement tenir compte des propriétés physico-chimiques du polymère à savoir sa viscosité minimale, sa comptabilité avec le renfort, sa masse moléculaire et sa mouillabilité. La prise en compte des propriétés mécaniques du polymère est également indispensable dans l’établissement d’une structure composite. Dans le cadre de la thèse, nous nous sommes intéressés au polysulfure de phénylène (PPS).
Le cas du polysulfure de phénylène Le polysulfure de phénylène (PPS) fait partie des polymères de hautes performances et thermostables utilisés notamment dans l’industrie aéronautique. Il fut pour la première fois synthétisé par Grenvesse en 1897. L’application industrielle de ce polymère avec la mise au point de plusieurs voies de synthèse détaillée par Lopez et Wilkes [Lamethe2004] est plus récente avec une première commercialisation dans les années 70. Il s’utilise principalement comme matériau non renforcé pour des applications dans les secteurs de l’automobile et de l’électronique. Il s’adapte également très bien en tant que matrice pour des pièces composites. Le PPS est un polymère semi-cristallin dont le monomère présente un cycle aromatique et un atome de soufre (S) représenté en [Figure 1.6]. Tout comme les matrices thermoplastiques, le PPS dispose d’une très bonne stabilité thermique. En raison d’une mobilité restreinte des chaînes du polymère imposée par le groupe phényle, la transition thermique du PPS se produit à haute température [Nohara+2006]. Ainsi le PPS conserve son intégrité structurelle (résistance thermique) jusqu’à une température de service de 200◦C en continu. Cette valeur est plus faible que celle déterminée pour le PEEK avec 240◦C [Villoutreix+1998]. Du fait de sa composition semi-cristalline, l’état du PPS se caractérise, à minima, par la température de transition vitreuse (Tg) et la température de fusion (Tm). La température de transition vitreuse, définissant la transition de l’état vitreux à l’état caoutchoutique de la phase cristalline, pour le PPS non renforcé est comprise entre 85◦C et 93◦C dans la littérature [Batista+2015 ; Biron2014 ; DíezPascual+2012]. Le point de fusion du PPS pur est estimé à 282◦C pour [Lamethe2004] et 285◦C selon [Batista+2015 ; Boey+1993 ; DíezPascual+2012 ; Nohara+2006]. Les différences constatées sont très certainement liées aux différents grades de PPS utilisés par les auteurs. Le PPS est un polymère adapté pour la mise en oeuvre de structures composites. Pour permettre l’imprégnation du renfort, la matrice doit être le plus fluide possible. Cela signifie qu’il est nécessaire d’atteindre la viscosité minimale du PPS. À l’état fondu, cette viscosité est d’environ 200 P a.s ce qui est faible par rapport aux autres thermoplastiques tels que le PEEK (∼ 350 P a.s). Des analyses rhéologiques [Figure 1.7] montrent que la viscosité du PPS à 315◦C varie peu pendant 30 min facilitant l’imprégnation dans le renfort et la cristallisation du PPS [Batista+2015]. L’établissement des propriétés mécaniques et thermiques du PPS est conditionné par la présence de la phase cristalline [Batista+2015]. Cette dernière améliore la rigidité et la résistance aux sollicitations mécaniques tandis que la phase amorphe participe à l’absorption d’énergie lors d’impacts. Un contrôle du ratio entre la phase cristalline et la phase amorphe lors de la mise en œuvre du matériau composite est primordial si l’on veut obtenir les performances mécaniques souhaitées. En partant de l’état fondu du PPS, la phase cristalline se forme lors de la phase de refroidissement du polymère. Ce changement thermique conduit à la formation de structures cristallines organisées et de formes sphériques appelé « sphérulites ». Chacune d’entre elles se développe radialement sous la forme de fines lamelles cristallines qui sont espacées par une phase amorphe. La morphologie et la répartition de ces sphérulites dépend fortement des propriétés physicochimiques et des conditions opératoires telles que la température de cristallisation, le taux de refroidissement ou encore la présence d’agents favorisant la nucléation des grains cristallins. Tout comme pour le PEEK ou le PEK, la cristallisation du PPS est un processus relativement long par rapport à des polymères conventionnels n’ayant pas de groupes aromatiques comme le polyéthylène (PE) [Nohara+2006]. La cinétique de cristallisation est fortement liée à la température de cristallisation. Une température trop proche de la transition vitreuse du PPS bloque le développement de la phase cristalline en raison d’une mobilité insuffisante des chaînes. Pour une température proche de Tm la cristallisation du PPS est également entravée. L’énergie disponible favorise la mobilité des chaînes sans pour autant permettre un réarrangement moléculaire propice à la cristallisation. C’est dans cette gamme de température proche de la transition vers l’état fondu du polymère que se produit une cristallisation secondaire caractérisée par des sphérulites incomplètes et de petites tailles. Les conditions en température permettant d’optimiser les cinétiques de nucléation et de croissance des grains cristallins sont donc comprises entre Tg et Tm [Spruiell2005]. Au cours de la mise en oeuvre, le PPS est particulièrement sensible à la vitesse de refroidissement appliquée. Cela se caractérise par une modification de la cinétique de cristallisation et le taux finale de la phase cristalline. Batista et al. [Batista+2015] montrent expérimentalement, pour un composite à matrice PPS renforcé par des fibres de verre, l’augmentation du taux de cristallisation lorsque le temps de refroidissement du polymère augmente [Tableau 1.1]. La même tendance se dégage dans la publication de [DíezPascual+2012] avec pour trois vitesses de refroidissement (2,0◦Cmin−1-5,0◦Cmin−1- 10,0◦Cmin−1) un taux de cristallinité respectif de 55,5%, 55,2% et 52,5% (cas du PPS FORTRON 0205B4 non renforcé). Dans le cadre de structures composites, le renfort influence les propriétés de la matrice au cours de la mise en œuvre. Dans les travaux de thèse de [Lamethe2004], un écart de la valeur de la température de transition vitreuse (Tg) d’au moins 10◦C entre un PPS non renforcé et un PPS renforcé fibres de carbone avec un taux massique de 60% a été constaté. La présence de fibres affecte la cinétique de cristallisation et le taux de cristallinité final pour une température donnée. Kenny et al. [Kenny+1991] démontrent, pour un cas de composite UD FC/PPS que la présence de ces fibres de renfort donne un taux maximal de cristallinité plus faible par rapport à une configuration PPS non renforcé. Néanmoins, la cinétique de cristallisation est plus rapide lorsque le PPS est en présence de fibres de carbone. Cette modification de comportement du PPS est à associer à la phase cristalline en contact avec le renfort. Au contraire, Auer et al. [Auer+1994] montrent expérimentalement que l’intégration de fibres de carbone ou de verre conduit à des cinétiques de cristallisation plus lentes [Figure 1.8]. C’est le cas contraire lorsque le polymère est renforcé de fibres d’aramide. Ces différences de résultats, notamment souligné par [Spruiell2005], peuvent s’expliquer par les spécifications et le traitement par ensimage ou non des fibres utilisées par les auteurs [Desio+1992]. Les fibres de renfort modifient le comportement de cristallisation de la matrice avec l’apparition d’une nouvelle phase cristalline appellée phase transcristalline [Figure 1.9]. Selon [López+1991], les fibres de carbone induisent une cristallisation particulière caractérisée par un nucléation plus efficace des germes cristallins à cette interface. L’écart des coefficients d’échange thermique entre le polymère et le renfort favorise la cristallisation du PPS en surface des fibres. L’auteur identifie également le poids moléculaire du polymère, le traitement des fibres ainsi que les paramètres de mise en oeuvre (vitesse de refroidissement) comme des facteurs supplémentaires pouvant favoriser la transcristallinité. Au final, l’intégration de fibres modifie la morphologie de la phase cristalline du PPS en particulier au niveau de l’interface fibre / matrice. Malgré un changement d’état réversible du PPS, plusieurs auteurs ont montré une dégradation rapide d’échantillons fondus à des températures supérieures à 300◦C lorsqu’ils sont soumis à une ambiance sous air. Cette dégradation est caractérisée lors d’un suivi cinétique avec l’inversion des modules de conservation G′ et de perte G′′ du matériau. Les premiers signes de dégradation surviennent dès 300◦C avec des réactions de branchements [Peters+1993]. À 320◦C, ce changement survient seulement au bout de 8 minutes [Lamethe2004]. Par ailleurs, la mise en fusion répétée du PPS entraîne, d’après Bulakh et al. [Bulakh+1993], une augmentation de sa réticulation rendant le matériau plus fragile. Les propriétés mécaniques du PPS dépendent à la fois de ses propriétés physico-chimiques, du procédé de mise en oeuvre et de son intéraction avec le renfort dans le cadre de fabrication de pièces composites. L’histoire thermique et le degré de cristallinité affecte significativement les propriétées mécaniques du PPS. Il existe plusieurs grades de PPS aux propriétés mécaniques variables. Le [Tableau 1.2] recense les propriétés mécaniques de plusieurs PPS commercialisés par les sociétés Celanese (FORTRON® ) et Toray (Torelina® ). Les différents grades disponibles de PPS diffèrent au niveau du poids moléculaire leur conférant ainsi des spécificités en terme d’utilisation et de choix de procédé de mise en oeuvre. Le PPS possède également une excellente résistance chimique y compris à haute température. Néanmoins, certains solvants usuels, de type alcalin ou acide, dégradent les propriétés mécaniques de ce polymère à haute température. L’association renfort/matrice constitue le semi-produit qui sera ensuite mis en œuvre pour obtenir la pièce composite finale. Plusieurs configurations de semi-produits sont disponibles suivant le type de pièce visé. Dans le cadre de la thèse, seuls les semi-produits thermoplastiques avec un renfort continu seront traités.
|
Table des matières
Introduction générale
1 État de l’art
1.1 Généralités sur les matériaux composites de la famille des thermoplastiques
1.1.1 Le choix du renfort et de la matrice
1.1.1.1 Le renfort : fibres de carbone
1.1.1.2 Les matrices thermoplastiques
1.1.1.3 Le cas du polysulfure de phénylène
1.1.2 Les semi-produits composites à matrice thermoplastique
1.1.2.1 Les architectures fibreuses tissées
1.1.2.2 Typologie des semi-produits thermoplastiques renforcés carbone
1.2 Effet de la mise en forme du semi-produit sur les propriétés de la pièce composite
1.2.1 Les problématiques liées à la mise en forme des préformes textiles
1.2.2 Les modes de déformation des préformes textiles
1.2.3 Les méthodes de caractérisation de la déformation en cisaillement des renforts textiles
1.2.3.1 Le « Bias Extension Test »
1.2.3.2 « Le Picture Frame »
1.2.3.3 Comparaison Bias extension Test – Picture Frame
1.2.4 Modélisation de la déformation d’une préforme textile
1.2.4.1 Approche cinématique
1.2.4.2 Vers la prise en compte du comportement mécanique du renfort
1.3 Caractérisation des structures composites par mesure sans contact
1.3.1 Mesure de champs par corrélation/stéréo-corrélation d’images
1.3.1.1 Principe de la corrélation d’image
1.3.1.2 Application aux renforts textiles
1.3.2 Contrôle non destructif de la santé matière du composite
1.3.2.1 Contrôle qualité d’une plaque composite par ultrason
1.3.2.2 Vers une analyse plus approfondie de la structure interne du matériau par tomographie à rayons X
1.4 Les procédés « classiques » de mise en œuvre par moulage de pièces composites
1.4.1 Généralités
1.4.2 Procédé retenu : la consolidation en autoclave
1.4.2.1 Présentation du dispositif
1.4.2.2 Le principe de la mise en œuvre par autoclave
1.4.2.3 Le choix des produits d’environnement
1.4.2.4 Définition du moule pour la consolidation de pièces à géométries complexes
1.5 La mise en œuvre des composites à matrice thermoplastique
1.5.1 Effet du procédé de consolidation par autoclave sur le matériau
1.5.2 Relation microstructure/propriétés mécaniques d’une pièce composite
1.6 Bilan intermédiaire
2 Mise en forme de préformes textiles sur des géométries à double courbures
2.1 Caractérisation en cisaillement plan par l’essai « Bias Extension Test »
2.1.1 Mise en place de l’essai « Bias Extension Test »
2.1.2 Comportement en déformation non-linéaire de la préforme textile
2.2 Étude de la drapabilité du semi-produit sur une géométrie complexe
2.2.1 Pilote d’emboutissage de préformes textiles et instrumentation
2.2.1.1 Présentation du pilote
2.2.1.2 Présentation du banc multi-caméras
2.2.1.3 Présentation du serre-flan
2.2.1.4 Présentation des poinçons d’emboutissage
2.2.1.5 Mise en place du renfort sur le dispositif de drapage
2.2.2 Mesure des champs de déformation du renfort textile
2.2.2.1 Validation du mouchetis artificielle pour mesurer les champs de déformation par stéréocorrélation d’images
2.2.2.2 Application de la technique de stéréocorrélation afin d’obtenir le champs de déformation
2.2.2.3 Calcul de l’angle de cisaillement
2.2.2.4 Validation du calcul de l’angle de cisaillement à travers l’essai « bias extension test »
2.2.3 Influence de la géométrie dans la déformation des préformes textiles
2.2.3.1 L’hémisphère
2.2.3.2 Tétraèdre
2.2.3.3 Poinçon parallélépipédique avec congés d’arête
2.2.4 Évaluation de la mesure du champ de déformation
2.3 Simulation du drapage à partir de la méthode du filet
2.3.1 Méthode
2.3.2 Hémisphère
2.3.3 Parallélépipède à congés d’arête
2.3.4 Tétraèdre
2.4 Bilan intermédiaire
3 Relations microstructure / propriétés mécaniques des plaques composites C/PPS
3.1 Les matériaux de l’étude
3.2 Mise en œuvre de plaques composites par consolidation autoclave
3.2.1 Description de la méthode de mise en oeuvre des plaques composites
3.2.2 Définition des cycles de consolidation
3.2.3 Les problématiques liées à la bâche à vide
3.2.3.1 Étanchéité de la bâche à vide
3.2.3.2 Incapacité du tissu drainant à remplir son rôle d’évacuation de l’air dans la poche à vide
3.2.4 Bilan des essais de consolidation
3.3 Les méthodes de caractérisation appliquées à l’analyse des plaques composites
3.3.1 Caractérisation mécanique des plaques composites
3.3.1.1 Traction sens chaîne et trame
3.3.1.2 Compression
3.3.1.3 Flexion 3 points
3.3.1.4 Cisaillement interlaminaire – CIL
3.3.2 Analyse microstructurale des stratifiés composites
3.3.2.1 Dissolution chimique
3.3.2.2 Analyse morphologie des porosités par CT-scan
3.4 Microstructure et propriétés mécaniques
3.4.1 Analyse de la microstructure
3.4.1.1 Plaque poudrée PORCHER
3.4.1.2 Plaque comélé SCHAPPE
3.4.1.3 Écart dans l’estimation du taux de porosité par dissolution chimique et par CT-scan
3.4.1.4 Comparaison des deux typologies de plaques
3.4.2 Résultats des essais mécaniques
3.5 Influence du cycle de consolidation sur les porosités et les propriétés mécaniques
3.5.1 Influence du cycle de consolidation sur la santé matière du composite
3.5.1.1 Effet de la pression externe
3.5.1.2 Effet du niveau de vide
3.5.1.3 Effet du temps de consolidation
3.5.1.4 Stratégie dans la définition du cycle autoclave
3.5.2 Sensibilité des propriétés mécaniques au taux volumique de porosité dans le stratifié
3.5.3 Prise en compte de la morphologie des porosités dans l’évolution des performances mécaniques
3.6 Bilan intermédiaire
4 Faisabilité de mise en œuvre de pièces composites à géométries complexes
4.1 Impact du cisaillement sur les propriétés mécaniques du matériau composite
4.1.1 Consolidation des plaques C/PPS avec déformation initiale en cisaillement plan
4.1.2 Effet du niveau de cisaillement plan sur les propriétés du matériau composite
4.1.2.1 Analyse microstructurale des plaques composites
4.1.2.2 Propriétés mécaniques des plaques composites
4.1.3 Confrontation avec une approche « théorie classique des stratifiés »
4.1.3.1 Détermination des propriétés élastiques du renfort textile
4.1.3.2 Détermination des propriétés élastiques du pli UD par méthode inverse
4.1.3.3 Modélisation du comportement mécaniques des plaques cisaillées
4.1.3.4 Prédiction de la rupture à partir du critère de Tsai-Hill
4.2 Application aux géométries complexes : consolidation de pièces à doublecourbures
4.2.1 Présentation des géométries
4.2.2 Mise en œuvre des pièces composites par consolidation autoclave
4.2.3 Évaluation de la santé matière des pièces complexes consolidées
4.2.3.1 Pièce de faisabilité : parallélépipède à congés d’arête
4.2.3.2 Pièce d’étude de la déformation textile : le tétraèdre
4.3 Bilan intermédiaire
Conclusion générale et perspectives
Annexes
Télécharger le rapport complet