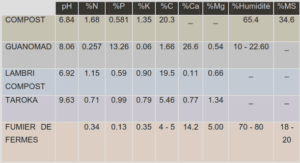
Télécharger le fichier pdf d’un mémoire de fin d’études
Pharmacovigilance
L’OMS définit la pharmacovigilance comme étant la science et les activités relatives à la détection, à l’évaluation, à la compréhension et à la prévention des effets indésirables ou de tout autre problème lié à l’utilisation des produits de santé [4].
Par « effet indésirable », on entend toute modification péjorative, avérée ou potentielle, de l’état de santé ou de bien-être d’un sujet traité, ou ayant été traité, par un médicament et qui peut être raisonnablement attribuée à ce dernier. Dans le cas où la relation de cause à effet n’est pas établie ou fortement suspectée, on doit, par convention, utiliser le terme d’« événement indésirable » [8]. Idéalement, la pharmacovigilance répond aux exigences successives : détecter le plus précocement possible un risque associé à un traitement médicamenteux, quantifier ce risque, le décrire (chez qui survient-il préférentiellement ? dans quelles conditions ?), si possible expliquer son mécanisme de survenue et, enfin, tenter de le prévenir par des mesures adaptées. La pharmacovigilance repose sur :
– Le recueil basé sur la notification spontanée des effets indésirables par les professionnels de santé, les patients et associations agréées de patients et les industriels avec l’appui du réseau des 31 centres régionaux de pharmacovigilance
– L’enregistrement et l’évaluation de ces informations
– La mise en place d’enquêtes ou d’études pour analyser les risques, la participation à la mise en place et au suivi des plans de gestion des risques
– L’appréciation du profil de sécurité d’emploi du médicament en fonction des données recueillies
– La prise de mesures correctives (précautions ou restriction d’emploi, contre-indications, voire retrait du produit) et la communication vers les professionnels de santé et le public
– La communication et la diffusion de toute information relative à la sécurité d’emploi du médicament
– La participation à la politique de santé publique de lutte contre l’iatrogénie médicamenteuse
Médicaments et autres produits de santé
Médicament
Selon l’article L 511 du code de la santé publique, on entend par médicament : «…toute substance ou composition présentée comme possédant des propriétés curatives ou préventives à des maladies humaines ou animales, ainsi que tout produit pouvant être administré à l’homme ou à l’animal en vue d’établir un diagnostic médical ou de restaurer, corriger ou modifier leurs fonctions organiques » [9].
Cette définition est maintenant remplacée par celle de l’article L.5111-1 du code de la Santé publique qui définit le médicament comme : « toute substance ou composition présentée comme possédant des propriétés curatives ou préventives à l’égard des maladies humaines ou animales, ainsi que toute substance ou composition pouvant être utilisée chez l’homme ou chez l’animal ou pouvant leur être administrée, en vue d’établir un diagnostic médical ou de restaurer, corriger ou modifier leurs fonctions physiologiques en exerçant une action pharmacologique, immunologique ou métabolique [10].
Les médicaments peuvent être classés en plusieurs catégories :
• Médicaments magistraux
Ce sont des médicaments préparés extemporanément à l’officine, en exécution d’une ordonnance émanant d’un praticien qualifié qui en précise la formule détaillée [39].
Cette formule est en principe originale et est adaptée au traitement d’un malade.
• Médicaments officinaux
Il s’agit d’une préparation dont la composition et le mode de préparation sont inscrits dans la pharmacopée ou dans un formulaire national [3].
• Produits officinaux divisés (POD)
Ce sont des produits fabriqués industriellement et figurant sur une liste établie par arrêté du ministre de la santé publique [3].
• Spécialités pharmaceutiques
Médicament préparé à l’avance, présenté sous un conditionnement particulier, caractérisé par une dénomination spéciale et vendu dans plus d’une officine [11].
• Substances vénéneuses
Les substances vénéneuses sont classées en deux sections.
La première section concerne les substances destinées à l’industrie où à l’agriculture, la deuxième celle destinées à la médecine.
A l’intérieur de chaque section, ces substances sont classées en trois tableaux :
Le tableau A (liste 1) est réservé aux produits toxiques Le tableau B est réservé aux produits stupéfiants
Le tableau C (liste 2) est réservé aux produits dangereux
• Les médicaments génériques
On entend par spécialité générique d’une autre spécialité, une spécialité qui a la même composition qualitative et quantitative en principes actifs, la même forme pharmaceutique et dont la bioéquivalence a été démontrée par des études appropriées de biodisponibilité ; les différentes formes pharmaceutiques orales à libération immédiate sont considérées ici comme une même forme pharmaceutique ” [12].
Le médicament générique obéit aux mêmes règles que le médicament princeps : mêmes procédures d’obtention de l’Autorisation de Mise sur le marché (AMM) (nationale ou européenne), mêmes principes et exigences permettant la démonstration de la qualité du médicament, sa reproductibilité d’un lot à l’autre et sa stabilité, mêmes règles de prescription et de délivrance.
Les obligations des fabricants et exploitants des médicaments génériques en matière de pharmacovigilance, de déclaration des effets indésirables, de gestion des risques et d’information sont identiques à celles des exploitants des médicaments de référence [7].
• Les médicaments essentiels
Le principe du médicament essentiel a été défini par l’OMS dans les années soixante-dix (70) pour répondre aux incertitudes d’approvisionnements des pays en développement : “ Les médicaments essentiels sont ceux qui satisfont aux besoins de la majorité de la population en matière de soins de santé ; ils doivent donc être disponibles à tout moment, en quantité suffisante et sous la forme pharmaceutique appropriée. ” [12].
Le médicament essentiel représente à la fois un concept thérapeutique (médicament possédant un ratio bénéfice/risque optimal, c’est-à-dire pouvant être utilisé dans de bonnes conditions de sécurité et un risque modéré et connu d’apparition d’effets secondaires) et économique en termes de santé publique. La liste des médicaments essentiels, composée de 400 à 450 molécules, régulièrement réévaluée par l’OMS, est une liste modèle destinée à aider les pays à identifier leurs propres priorités et à faire leur propre sélection. Une liste de médicaments essentiels doit pouvoir régler la plupart (80 à90%) des problèmes de santé qui nécessitent un traitement dans une population dans des conditions normales [12].
• Les médicaments sous standards
Les médicaments sous-standards, bien que n’ayant pas de définition juridique également appelés « produits hors spécification « OOS – out-of-spécification », sont des médicaments authentiques produits par des fabricants autorisés par l’autorité nationale de réglementation qui ne remplissent pas les spécifications de qualité élaborées dans le cadre national pour ces produits [13]. Normalement, chaque médicament produit par un fabricant doit respecter les standards de qualité et les spécifications associées. Ces standards et spécifications sont analysés et évalués par l’autorité nationale de réglementation avant que le produit ne soit autorisé pour la commercialisation (AMM).
Le terme « non conforme» signifie quant à lui qu’un médicament présente certains écarts par rapport aux spécifications requises par le dossier d’enregistrement approuvé par l’autorité d’enregistrement et/ou aux bonnes pratiques de fabrication en vigueur dans le pays d’enregistrement.
Sur le terrain, ces médicaments sous-standards ou non conformes pouvant être sous-dosés ou mal étiquetés, posent de réels problèmes de santé publique [13].
Autres produits de santé
Ils incluent les médicaments et les produits contraceptifs, le sang et ses dérivés, les vaccins, les produits biologiques, les produits de la biotechnologie, les produits diététiques et les suppléments alimentaires, les plantes médicinales, les produits cosmétologiques, les produits homéopathiques ainsi que les produits utilisés dans le diagnostic biologique et ainsi que les produits utilisés dans le diagnostic biologique et radiologique et les produits à usage vétérinaire que ces produits aient bénéficié de l’autorisation de mise sur le marché ou qu’ils soient en cours de développement [14].
Objectifs de la pharmacovigilance
Des événements tels que la tragédie de la thalidomide font ressortir l’extrême importance de systèmes efficaces de contrôle des médicaments. Les principaux objectifs des programmes de pharmacovigilance sont :
– De détecter précocement les effets et interactions indésirables nouveaux
– De détecter les augmentations de fréquence des effets indésirables connus
– D’identifier des facteurs de risque et des mécanismes pouvant expliquer les effets indésirables ;
– D’évaluer le rapport bénéfice/risque et la diffusion de l’information nécessaire ;
– D’améliorer la prescription et la réglementation du médicament. Le but final de la pharmacovigilance est :
– L’utilisation rationnelle et en toute sécurité du médicament,
– L’évaluation et la communication du rapport bénéfice/risque des médicaments mis sur le marché,
– L’éducation et l’information des patients.
Terminologie de base
• Effet secondaire
Un effet secondaire est un effet survenant en plus de l’effet principal lors de la prise d’un traitement médicamenteux [15].
Prenons un exemple : le médecin prescrit un traitement pour soigner une bronchite. L’effet principal est la diminution de la toux, de la gêne respiratoire et des douleurs. L’effet secondaire peut-être l’augmentation de l’envie de dormir car la molécule chimique contenue dans le médicament a également une action sur le système nerveux qui commande l’éveil et le sommeil.
La liste des effets secondaires est inscrite sur la notice du médicament. Cependant ces effets secondaires ne se produisent pas systématiquement. Et cette liste ne signifie pas qu’ils vont tous avoir lieu. Ils figurent sur la notice surtout pour prévenir qu’ils peuvent se produire.
Tous les types de traitement peuvent entraîner des effets secondaires, chacun réagit de façon différente aux médicaments ce qui veut dire qu’ils ne sont pas toujours nocifs. Pour certains les effets secondaires seront légers et peu gênants, pour d’autres ils seront difficilement supportables, en particulier quand cet effet secondaire devient un effet indésirable. De nombreux effets indésirables sont bénins. Cependant, certains effets secondaires indésirables peuvent être sévères. Il convient ainsi de mesurer le rapport bénéfice/risque qui évalue l’intérêt du médicament en comparaison avec les effets secondaires indésirables qu’il peut présenter.
• Effet indésirable
Une réaction nocive et non voulue, se produisant aux posologies normalement utilisées chez l’homme pour la prophylaxie, le diagnostic ou le traitement d’une maladie ou la modification d’une fonction physiologique [16]. C’est également toute réaction résultant d’un (e) :
– Mésusage
– Usage abusif
– Syndrome de sevrage
– Pharmaco dépendance
– Erreur médicamenteuse
– Inefficacité thérapeutique
– Effet sur le produit de conception
– Produit défectueux ou de mauvaise qualité
– Iatrogénie médicamenteuse
• Effet indésirable grave
Effet indésirable létal ou susceptible de mettre la vie en danger ou entrainant une invalidité ou une incapacité ou provoquant ou prolongeant une hospitalisation ou se manifestant par une anomalie ou malformation.
• Effet indésirable inattendu
Effet indésirable dont la nature, la sévérité/intensité ou l’évolution ne correspondent pas aux informations contenues dans le résumé des caractéristiques du produit.
• Évènement indésirable
Manifestation nocive et non recherchée, survenant chez un sujet pendant un traitement. Le terme évènement indésirable contrairement à effet indésirable ne préjuge pas d’un lien causal avec une exposition notamment à un médicament.
La pharmacovigilance internationale, Rôle d l’OMS
A l’échelle mondiale, la pharmacovigilance dépend de la puissance de systèmes nationaux qui contrôle la mise au point et la qualité des médicaments, notifient leurs effets nocifs, et fournissent des informations exactes pour les utiliser sans danger. Le Sénégal échange des données sur la pharmacovigilance avec l’ensemble des pays ayant adhéré au programme international de pharmacovigilance. En effet, le Sénégal est devenu, depuis le 26juin 2009, pays membre à part entière de ce programme, devenant ainsi le 95ème pays membre au plan mondial et le 2ème pays en Afrique noire francophone à avoir accédé à ce stade. La direction de la pharmacie et du médicament échange des informations avec le centre collaborateur de l’OMS, basée à Uppsala (suède), UMC, qui héberge la base de données mondiale des effets indésirables médicamenteux. L’UMC est une fondation indépendante et un centre de service international et de la recherche scientifique. Il vise à améliorer la sécurité des patients dans le monde entier. Ses priorités sont la sécurité des patients et l’utilisation sure et efficace des médicaments dans chaque partie du monde. Il concentre ses propriétés en recherche et développement innovant, en fournissant des données de référence aux organismes de règlementation, aux professionnels de santé, aux chercheurs et à l’industrie pharmaceutique dans le monde entier [14].
Il vise un monde où tous les patients et les professionnels de santé vont prendre des décisions thérapeutiques sages lors de l’utilisation des médicaments.
Le but ultime de ce centre travaillant dans la pharmacovigilance est de soutenir la bonne prise de décision concernant les avantages et le risques des options de traitement pour les patients prenant des médicaments. En effet, il contribue à faire :
– Le dépistage et l’analyse de données internationales sur les effets indésirables ; son objectif est de détecter, des problèmes potentiels, importants, chez les patients et sur la santé publique concernant l’utilisation et la sécurité des médicaments
– Soutenir la communication efficace, la mise à jour des informations scientifiques.
– Fournir des outils pour la saisie de données, la gestion et la recherche
– L’éducation et la formation dans la mise en place et l’exécution des programmes nationaux de pharmacovigilance, ainsi que dans l’utilisation des outils de l’UMC. L’existence d’un réseau internationale de surveillance des effets indésirables des médicaments comme celui de l’OMS peut fournir des informations importantes sur la sécurité d’utilisation des médicaments avant que ces informations ne soient générées par les banques de données nationales. La pharmacovigilance est nécessaire pour prévenir des pathologies induites par l’utilisation des médicaments et pur réduire les conséquences économiques liées aux effets indésirables [14].
Dans le cadre de surveiller les effets indésirables des médicaments et d’harmoniser le système de pharmacovigilance, l’OMS mis en place un programme de pharmacovigilance international lancé depuis 1968 mettant en commun les données existantes sur les réactions indésirables aux médicaments. Il s’agissait à l’origine d’un projet pilote mené dans dix pays avec les systèmes nationaux existants de notification des effets indésirables. Depuis, ce réseau a été sensiblement élargi à mesure que davantage de pays dans le monde mettaient en place des centres nationaux de pharmacovigilance pour l’enregistrement des réactions indésirables aux médicaments. Actuellement, 86 pays participent à ce programme qui est coordonné par l’OMS avec l’aide de son centre collaborateur d’Uppsala, en Suède. Le centre collaborateur se charge d’alimenter la base de données mondiale (Vigibase) sur les réactions indésirables aux médicaments. Actuellement, cette base de données contient plus de 3 millions de notifications de réactions indésirables.
Le centre collaborateur de l’OMS analyse les notifications arrivant dans la base de données afin :
– De recenser précocement les signaux d’alerte concernant des réactions indésirables graves à des médicaments
– D’évaluer le risque
– D’entreprendre des recherches sur les mécanismes pouvant aider à mettre au point des médicaments plus sûrs et plus efficaces [30].
L’OMS, à travers un comité consultatif, joue un rôle important dans la fourniture d’avis d’experts sur toutes les questions relatives à la sécurité et à l’innocuité des médicaments. Ce comité se charge aussi de promouvoir l’adoption de politiques et de lignes d’action cohérentes entre les États Membres et de renseigner les entités concernées sur les mesures prises dans d’autres pays.
Le succès du Programme de pharmacovigilance internationale de l’OMS dépend entièrement des apports des centres nationaux de pharmacovigilance. Ces centres constituent un pôle essentiel d’expérience et de compétence qui a joué un rôle actif dans le développement continu du programme de l’OMS et de la pharmacovigilance dans son ensemble. Idéalement, chaque pays devrait être doté d’un centre de pharmacovigilance [33].
Les données collectées dans un pays ou dans une région sont d’une grande pertinence et d’une valeur éducative indéniable, et peuvent aider les organismes de réglementation dans la prise de décision. En effet, l’information obtenue dans un pays donné (par exemple le pays d’origine du médicament) peut ne pas être adaptée à d’autres régions du monde où les circonstances d’utilisation peuvent différer. Par ailleurs, la non-disponibilité de l’information dans une région donnée peut retarder l’alerte des autorités de réglementation, des médecins, des pharmaciens, des patients et de l’industrie pharmaceutique [34].
L’existence d’un réseau international de surveillance des effets indésirables des médicaments comme celui de l’OMS peut fournir des Informations importantes sur la sécurité d’utilisation des médicaments avant que ces informations ne soient générées par les banques de données nationales. La pharmacovigilance est nécessaire pour prévenir des pathologies induites par l’utilisation des médicaments et pour réduire les conséquences économiques liées aux effets indésirables. En conclusion, les médicaments mis sur le marché nécessitent une surveillance continue dans chaque pays [34].
Système nationale de pharmacovigilance
Organisation du système
Le Système National de Pharmacovigilance est composé de l’ensemble des structures publiques et privées, et de toutes les personnes physiques qui interviennent dans les activités de pharmacovigilance. Parmi ces structures on peut citer :
• Au niveau central la DPM (Direction de la Pharmacie et du Médicament), le Centre Antipoison (CAP), le Laboratoire Nationale de Contrôle des Médicaments (LNCM), la Pharmacie Nationale d’Approvisionnement (PNA) et les programmes de santé.
• Au niveau intermédiaire (la Région Médicale)
• Au niveau périphérique (les districts et les Postes de santé, cabinets médicaux, pharmacies…) [35]
Structures et acteurs du système de pharmacovigilance
Ils sont portés par l’article 6 de l’arrêté ministériel n° 05036 du 22 avril 2009.
La direction de la pharmacie et du médicament DPM
Elle définit les orientations en matière de pharmacovigilance, veille au respect des procédures de surveillance et coordonne les activités du SNPV. Elle reçoit les déclarations, rapports et toutes les informations qui lui sont transmis. Le Directeur de la Pharmacie et du médicament convoque les réunions de la Commission Nationale de Pharmacovigilance. Elle peut faire effectuer tous travaux ou enquêtes nécessaires et prendre, après exploitation des informations, toutes mesures appropriées ou saisir les autorités compétentes. La DPM est en relation avec les institutions de la pharmacovigilance internationale [35].
La DPM est ainsi l’autorité nationale de réglementation. Ses missions émanant du décret Selon le décret N° 2004-1404, du 04 novembre 2004 portant organisation du Ministère de la Santé et de la Prévention Médicale, sont :
– D’élaborer et veiller à l’application des textes législatifs et réglementaires relatifs à la pharmacie, aux médicaments, aux réactifs de laboratoires d’analyses médicales, aux substances vénéneuses, à l’alcool et aux dispositifs médicaux
– De réglementer l’exercice des professions pharmaceutiques et contrôler les laboratoires d’analyses médicales
– De réglementer et d’assurer la promotion de la pharmacopée traditionnelle.
La DPM comprend : [36]
– La Division des études, de la réglementation et de la documentation
– La Division du contrôle administratif des médicaments
– La Division des stupéfiants et des substances psychotropes
– La Division des Laboratoires d’analyses médicales
– Le Bureau de Gestion.
Il convient de doter toutes les divisions et les bureaux de la DPM de personnels et d’assurer leur formation et leur perfectionnement pour une meilleure efficacité dans le travail. La DPM a aussi besoin de personnel d’appui secrétaires, chauffeurs, agents pour le nettoiement et l’acheminement du courrier.
Le problème crucial de la DPM réside dans le manque de moyens pour une exécution correcte de ses activités. Ainsi, la DPM manque−t−elle de personnel, d’ordinateurs, de véhicules, d’accès à Internet et de bien d’autres équipements logistiques essentiels ? [37].
La commission nationale de pharmacovigilance (CNPV)
La CNPV est une instance officielle consultative, chargée de soumettre à la décision du Ministre, des mesures pour prévenir ou arrêter des incidents liés à l’utilisation des médicaments. La CNPV peut proposer au Ministre de la Santé les enquêtes et les travaux qu’elle estime utiles à l’exercice de la pharmacovigilance. Elle se réunit au moins deux fois par an et chaque fois que de besoin, sur convocation de son président. Sauf cas d’urgence, les travaux de la CNPV sont préparés par le Comité Technique [38].
Le comité technique de pharmacovigilance
Le Comité Technique prépare les travaux de la Commission Nationale de Pharmacovigilance. Il est chargé :
– De valider les résultats d’imputabilité des cas d’effets indésirables déclarés
– D’évaluer les risques médicamenteux encourus par la population
– De coordonner, recenser, évaluer des enquêtes et travaux de pharmacovigilance.
Il se réunit au moins deux fois par an et chaque fois que de besoin sur convocation de son Président [31].
Au sens de l’article 12 l’arrêté 0536 du 22 avril 2009, c’est un comité d’expert avec un pharmacologue, un toxicologue, un épidémiologiste, un neurologue, un dermatologue, un pédiatre et un immunologiste [38].
|
Table des matières
INTRODUCTION
PREMIERE PARTIE
1. Généralités sur la pharmacovigilance
1.1. Historique
1.2. Définitions
1.2.1. Pharmacovigilance
1.2.2. Médicaments et autres produits de santé
1.2.2.1.Médicament
1.2.2.2.Autres produits de santé
1.3. Objectifs de la pharmacovigilance
1.4. Terminologie de base
1.5. Champ d’application de la pharmacovigilance
1.6. Bases réglementaires
1.7. Approche méthodologique
1.8. Intérêt de la pharmacovigilance
1.9. La pharmacovigilance internationale, Rôle d l’OMS
2. Système nationale de pharmacovigilance
2.1. Organisation du système
2.2. Structures et acteurs du système de pharmacovigilance
2.2.1. La direction de la pharmacie et du médicament DPM
2.2.2. La commission nationale de pharmacovigilance
2.2.3. Le comité technique de pharmacovigilance
2.2.4. Le centre antipoison
2.2.5. Le laboratoire national de contrôle de qualité des médicaments
2.2.6. Les hôpitaux
2.2.7. La région médicale
2.2.8. Les laboratoires pharmaceutiques
2.2.10. Les programmes de santé
2.2.11. L’OMS et les autres partenaires
2.2.12. Les autres acteurs
2.3. Processus de transmission de la notification
2.3.1. La notification
2.3.2. Transmission et circuit de la fiche de notification
2.3.3. Traitement des données et prise de décision
2.3.3.1. L’imputabilité
2.3.3.2. La prise de décision
2.3.3.3. La retro information ou feedback
2.4. Procédure d’enquête et de suivi de pharmacovigilance
3. Fardeau des effets indésirables médicamenteux
3.1. Fardeau en termes de morbidité et de mortalité
3.2. Fardeau économique
1. Généralités sur la Qualité des médicaments
1.1. Définition
1.2. Notion de qualité, sécurité, efficacité
1.3. Assurance qualité, Bonnes Pratique de Fabrication (BPF), Contrôle de qualité
2. Critères de qualité d’un médicament
3. Qualité des matières premières
3.1. Le principe actif
3.2. L’excipient
3.3. Le récipient
4. Cas des médicaments de qualité inférieur et falsifiés
4.1. La contrefaçon
4.2. Nouvelles définitions des médicaments de qualité inférieure ou falsifiés
4.3. Principaux faits
4.5. Ampleur du problème
4.6. Comment traiter ce problème
DEUXIEME PARTIE : TRAVAIL PERSONNEL
1. But
2. Objectifs
2.1. Objectif général
2.2. Objectifs spécifiques
3. Cadre d’étude
3.1. Le centre Hospitalier universitaire de Fann
3.2. L’Hôpital Principal de Dakar
3.3. Centre Hospitalier National Abass Ndao
3.4. Centre hospitalier universitaire Aristide le Dantec
4. Méthodologie
4.1. Type et période d’étude
4.2. Population d’étude
4.3. Échantillonnage
4.3.1. Critères d’éligibilité
4.3.2. Taille de l’échantillon
4.3.3. Procédure de sélection
4.4. Collecte des données
4.4.1. Outil de collecte
4.4.2. Méthode de collecte
4.4.3. Variables collectées
4.4.4. Définition opérationnelle des variables
4.5. Analyses statistiques
4.6. Considérations éthiques
5. Résultats
5.1. Taux de réponse
5.2.1.Age
5.2.2.Sexe
5.2.3.Statut professionnel
5.2.4.Service
5.2.5.Expérience professionnelle
5.2.6.Formation sur la pharmacovigilance
5.2.7.Nombre de réunion/rencontre sur la pharmacovigilance au cours des deux dernières années
5.3. Attitudes
5.4. Efficacité, toxicité, défaut de qualité, tolérance
5.4.1. Efficacité
5.4.1.1. Avez-vous déjà vu le formulaire de notification de pharmacovigilance en vigueur au Sénégal
5.4.1.2. Avez-vous été confronté à un problème d’efficacité de médicaments chez les patients
5.4.1.3. Le nombre de fois confronté à un problème d’efficacité de médicaments chez les patients
5.4.1.4. Si oui, pouvez- vous citer un (des) exemple (s) de médicaments en cause
5.4.1.5. Signalement au moins une fois du problème d’efficacité
5.4.1.6. Lieu de signalement de problème d’efficacité
5.4.1.7. Après l’avoir signalé, avez-vous eu un accusé de réception ?
5.4.1.8. Après l’avoir signalé, avez-vous eu une rétro information ?
5.4.1.9. Si vous ne l’avez pas signalé, veuillez indiquer la ou les raisons
5.4.2. Toxicités
5.4.2.1. Problème de toxicité de médicaments chez les patients
5.4.2.2. Citez un exemple de médicaments en cause
5.4.2.3. Signalement au moins une fois du problème de toxicité
5.4.2.5. Réception d’un accusé de réception
5.4.2.6. Réception d’une rétro information
5.4.2.7. Raisons de non signalement
5.4.3. Défaut de qualité
5.4.3.1. Problème de défaut de qualité de médicaments
5.4.3.2. Les médicaments en cause du défaut de qualité
5.4.3.3. Signalement du défaut de qualité de médicaments
5.4.3.4. Lieu de signalement de problème de défaut de qualité de médicament..
5.4.3.5. Reçu un accusé de réception
5.4.3.6. Reçu un feedback
5.4.3.7. Raisons de non signalement
5.4.4. Tolérance
5.4.4.1. Constater des EIM
5.4.4.2. Nombre de fois des EIM
5.4.4.3. Les mesures prises pour prendre en charge l’EIM
5.4.4.4. Signalement au moins une fois des EIM
5.4.4.5. Lieu de signalement des EIMs
5.4.4.6. Reçu un accusé de réception
5.4.4.7. Reçu un feedback
5.4.4.8. Raisons de non signalement
5.5. Promotion de la pharmacovigilance
5.6. Obstacles au signalement des effets indésirables médicamenteux
5.7. Facteurs susceptibles de motiver les pharmaciens et prescripteurs à signaler les effets indésirables médicamenteux.
5.8. Sources d’informations
DISCUSSION
1. Limites de l’étude
2. Caractéristiques socio-professionnelles
4. Pratique envers la pharmacovigilance
4.1. Surveillance des médicaments
4.2. Promotion de la pharmacovigilance
5. Facteurs facilitants
6. Facteurs limitants
7. Sources d’information
CONCLUSION ET RECOMMANDATIONS
RÉFÉRENCES BIBLIOGRAPHIQUES
ANNEXES
Télécharger le rapport complet