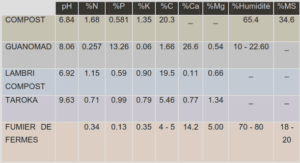
Télécharger le fichier pdf d’un mémoire de fin d’études
Les réseaux permettent de visualiser les données relationnelles
« Une image vaut mille mots » (Confucius)
Le dessin d’un réseau permet d’appréhender de façon intuitive des données relationnelles complexes. Il permet de voir un jeu de données, de distinguer rapidement les caractéristiques clés d’un phénomène, de construire des hypothèses. Les représentations graphiques sont plus intuitives que les tableaux de données parce que notre cerveau a davantage été sélectionné au cours de son évolution pour la représentation mentale des formes que pour le calcul matriciel. La richesse des formes d’un réseau sollicite à plein ses capacités visuelles. Les réseaux sont des objets mathématiques faciles d’accès, car les concepts, mesures et algorithmes de réseaux sont souvent imagés.
La figure 1-2 n’est qu’une étape préliminaire pour qui souhaiterait analyser en finesse la structure sociale du groupe de Roscoff. Ce dessin représente la topologie du réseau, c’est-àdire les connexions entre sommets. Il est possible de mettre en valeur certains aspects de cette topologie en modifiant des aspects graphiques tels que la couleur, la taille ou la forme des noeuds et arêtes. Si l’on s’intéresse par exemple à la variation du nombre de connaissances des individus, on peut représenter le degré des noeuds par un code couleur (Figure 1-3.A). Cela fait ressortir certains individus isolés et d’autres très connectés.
L’individu 5 est très fortement connecté avec 16 voisins. Il occupe donc une place importante dans ce réseau social, ce qui n’est pas surprenant étant donné qu’il s’agit de Gaëlle Boutin, gestionnaire de l’école doctorale. On peut s’intéresser à des questions plus avancées, comme la circulation d’informations via les individus dans ce réseau. Une façon d’identifier les noeuds de communication clés est de représenter leur centralité d’intermédiarité (betweenness), à savoir la proportion de plus courts chemins entre individus dans le réseau qui passent par un sommet donné (Figure 1-3.B). En plus du sommet 5 qui est très central selon cette mesure, d’autres sommets plus périphériques ressortent car ils créent des liens vers des parties isolées du réseau.
Les réseaux sont adaptés à une démarche exploratoire
Ces quelques exemples montrent que la représentation de données par un réseau permet d’adopter naturellement une démarche exploratoire. La visualisation fait rapidement naître des hypothèses, guidée par des données qui n’avaient pas nécessairement été collectées pour y répondre. L’exploration est facilitée par la flexibilité du réseau, qui peut être associé à d’autres informations, voire redéfini différemment, pour approfondir les questions soulevées. Cette méthode exploratoire, distincte et complémentaire de la méthode hypothético-déductive [Kell, Oliver, 2004], est très pertinente pour s’intéresser à des objets nouveaux ou peu connus, tel que nous le ferons avec des séquences génétiques dans ce travail de thèse.
Les représentations graphiques sont des guides très utiles pour s’approprier les données, mais la démarche exploratoire ne doit pas s’y arrêter. Il serait hâtif de tirer des conclusions sur la structure du groupe de Roscoff à partir des dessins de la figure 1-3. Il est en effet difficile d’appréhender tous les détails de la structure d’un réseau en observant sa projection en 2 dimensions, ce qui est d’autant plus vrai que la taille du réseau augmente. Une solution serait de visualiser des parties restreintes du réseau, ou de comparer des projections selon plusieurs méthodes de placement des noeuds et arêtes, mais cette démarche est subjective, non systématique, et risque donc d’être biaisée.
Les doctorants de première année sur la figure 1-3.C sont ainsi décrits comme « périphériques », alors que certains ont pourtant un nombre élevé de connaissances (p. ex. les 11 et 12) : ils ne se retrouvent en périphérie que par le hasard de l’algorithme de projection. Pour aller au bout de la démarche, il s’agirait maintenant de formaliser en terme de réseau ce qu’est un noeud périphérique, en s’appuyant par exemple sur le riche corpus de concepts et d’algorithmes de la théorie des graphes. C’est dans ce cycle d’allers-retours entre représentations, construction d’hypothèses, formalisation et mesures de propriétés que la démarche exploratoire prend tout son intérêt.
Les réseaux permettent l’étude statistique des données relationnelles
A travers l’étude d’un groupe social, nous avons montré que les réseaux permettent de manipuler facilement des informations complexes, et qu’ils sont adaptés pour adopter une démarche exploratoire. Au-delà de ces aspects pratiques, le principal intérêt des réseaux est sans doute qu’ils permettent de poser un regard global sur les phénomènes, de les aborder comme des systèmes, à la différence des méthodes réductionnistes, plus classiques en science. Les réseaux proposent en fait de procéder à un type nouveau de statistiques, qui traite la structure des données relationnelles [Brandes et al., 2013].
Les statistiques sont l’étude des données : elles s’intéressent à la collecte, au traitement, à l’interprétation et la présentation des données, afin de les rendre intelligibles. Les études statistiques habituelles sont conçues pour des ensembles sans structure, des unités ou des groupes indépendants. Puisque les phénomènes réels présentent généralement une structure, une grosse partie du travail de statistiques consiste à détecter les relations entre données, pour éliminer les biais de dépendance. Si l’on étudie par exemple l’association entre le nombre d’amis d’un doctorant et son année de thèse, on cherchera à échantillonner des doctorants qui ne sont pas amis les uns des autres. Certaines études statistiques classiques s’intéressent à des données relationnelles, comme par exemple la corrélation entre les âges de paires d’amis, mais elles considéreront alors des paires indépendantes, sans individu en commun.
Les réseaux proposent au contraire de s’intéresser à la structure des données relationnelles. Cette approche repose sur l’hypothèse, implicite mais très forte, que la structure des relations est fondamentalement importante. Elle pense cette structure comme une propriété émergente du système étudié, qui n’est pas résumée par la simple agrégation de ses éléments constitutifs. C’est cette conceptualisation qui permet, et nécessite, de nouvelles méthodes de pensée, une forme nouvelle d’inférence scientifique et le développement de nouveaux types de connaissances. Nous allons exploiter cet avantage en réalisant des réseaux de similarité de séquences.
Les scores d’alignements reposent sur un modèle d’évolution
De nombreuses méthodes existent pour comparer les textes des séquences génétiques.
On peut comparer des descriptions globales comme leur longueur, la fréquence de certains mots, la proportion des différentes lettres. Ces méthodes sont employées pour des questions particulières, mais l’on procède plus habituellement à des alignements à des mises en correspondance entre les lettres le long des paires de séquences à comparer. L’hypothèse sous-jacente à un alignement est que les textes des séquences ont une origine ancestrale commune, et que leurs différences proviennent des générations de copies imparfaites qui les séparent. On imagine que certains mots ont pu être écrits différemment par les copistes ou remplacés par des synonymes, que des mots ou paragraphes ont pu être ajoutés ou supprimés. Pour rechercher les traces de ces liens ancestraux entre les textes observés aujourd’hui, on modélise leur évolution passée. On suppose qu’il existe des probabilités fixes que telle lettre ait été remplacée par telle autre, ou qu’un fragment de taille donnée ait été ajouté ou retiré. Ce modèle se traduit par un score que l’on attribue à l’alignement d’une paire de séquences : des correspondances entre fragments identiques font monter ce score, des correspondances entre lettres facilement interchangeables sont moins valorisées, tandis que des correspondances entre lettres très différentes, ou avec des trous insérés dans l’autre séquence font baisser le score. En pratique les paramètres exacts des matrices de similarité entre lettres sont calibrés sur des jeux de séquences dont on connaît l’origine ancestrale commune. On peut utiliser différentes matrices calibrées selon différentes méthodes (p. ex. BLOSUM, PAM, WAG) et sur des jeux de divergence variable (p. ex. BLOSUM 45, 62, 80 pour des séquences avec moins de 45, 62 ou 80% de résidus identiques). La valeur négative d’un trou (gap) est souvent modélisée par une fonction affine de sa taille : un coût à l’ouverture, un coût à l’extension. In fine, on obtient une formule mathématique qui associe un score à l’alignement d’une paire de séquences.
Système de score employé par BLAST
Le calcul du score brut d’alignement de BLAST S dépend de la matrice de similarité et du coût d’ouverture des gaps employés, tel qu’exposé dans la partie 1.3.1. Tel quel, il n’est pas comparable entre recherches et ne permet pas d’estimer la signification statistique d’un HSP. Ce score est donc normalisé en étudiant la distribution des scores de HSP entre des protéines aléatoires. Le modèle de protéines aléatoires employé consiste simplement à tirer chaque acide aminé indépendamment, selon la distribution moyenne dans les séquences [Altschul, Gish, 1996]. Les scores bruts des HSP entre de telles protéines aléatoires suivent alors une loi de distribution connue, caractérisée par la longueur des protéines m et n et les caractéristiques de la recherche1. Il est ainsi possible de calculer la probabilité d’obtenir un score au moins aussi bon « au hasard » (P-value), selon ce modèle de protéine aléatoire. La qualité d’un HSP est généralement décrite par sa E-value E, définie comme le nombre de HSP avec un score au moins aussi bon que S attendus (Expected) pour une recherche entre 2 protéines aléatoires de mêmes tailles. La E-value est donnée par la formule E = Km*n e-λS. Elle augmente proportionnellement à la taille des séquences comparées, car dans une séquence aléatoire deux fois plus longue, on peut s’attendre à trouver 2 fois plus de HSP avec un score supérieur à S. La E-value décroit exponentiellement avec le score S.
Cette décroissance exponentielle se comprend mieux en termes de probabilité2. Un HSP de score S1+S2 correspond à deux HSP consécutifs de scores S1 et S2, sa probabilité est donc le produit des probabilités de HSP de score S1 et S23. Cette multiplication des sous-scores explique la décroissance exponentielle de la E-value avec le score (car e-λS1 * e-λS2 = e-λ(S1+S2)).
En pratique, le score d’alignement est le déterminant principal de la E-value.
Une recherche d’alignements locaux par BLAST s’effectue habituellement contre une base de données cible comprenant plusieurs séquences. Pour calculer la E-value, BLAST considère la base de donnée comme une longue séquence de taille n. La E-value d’une HSP doit donc se comprendre dans son contexte, c’est-à-dire dans le cadre d’une recherche contre une base de donnée d’une certaine taille. Une autre transformation du score brut est fournie par BLAST. Il s’agit du bit score1 S’ qui permet des comparaisons entre recherches effectuées avec différents systèmes de score, sans prendre en considération la longueur des séquences comparées. Ce score est exprimé dans une échelle logarithmique de base 2, d’où le nom bit qui réfère à une unité d’information. Une augmentation de 1 du bit score correspond à une division par 2 de la E-value.
Le bit-score est plus pratique que la E-value à plusieurs égards. Tout d’abord, il augmente avec la qualité d’une HSP tandis que la E-value décroit de façon contre-intuitive. Le bit score est généralement compris entre 0 et quelques centaines. Il s’approxime donc par un chiffre entier, tandis que la E-value s’exprime entre 0 et 1 et s’approxime par une puissance négative de 10. Si la variation exponentielle de la E-value avec le score à un sens statistique contre un modèle de protéine aléatoire, la variation du bit score est plus naturelle pour comparer la qualité de deux HSP. Enfin le bit score dépend uniquement de la qualité de la HSP, et pas de la longueur des séquences comparées, ce qui permet de comparer des HSP obtenus indifféremment contre des petites ou des grandes bases de données. De plus il n’est pas forcément pertinent de réduire la valeur attribuée à une HSP selon qu’elle ait été obtenue dans une petite ou dans une grande séquence. Malgré cela, l’usage dans la littérature est d’employer la E-value comme échelle de référence pour les recherches BLAST. On utilise parfois la transformation -log(E-value), qui a les premiers avantages du bit score, mais qui dépend de la longueur des séquences comparées.
Le choix d’un seuil sur la E-value n’a pas de sens statistique
La E-value (nombre attendu de HSP aléatoires de score >= S) ne devrait a priori pas être confondue avec la P-value (probabilité d’obtenir une HSP aléatoire de score >= S). Mais ces notions sont en fait très proches, et sont quasiment égales pour les petites valeurs qui nous intéressent (<0,1)2. Si l’on raisonnait en termes statistique habituels, on emploierait donc un seuil de 0,05 lors de l’alignement d’une séquence requête contre une base de données, pour limiter les chances d’obtenir des HSP dus à la pure variation aléatoire (1 chance sur 20). En pratique, les seuils employés sont beaucoup plus restrictifs. On considère rarement qu’un HSP est de bonne qualité lorsque sa E-value est supérieure à 10-5. Des seuils encore plus restrictifs sont même souvent employés (10-10 voire 10-20), et d’autres indicateurs de la qualité d’une HSP sont considérés en complément, en particulier le pourcentage de résidus identiques dans l’alignement. Pourquoi utiliser des seuils de E-value si stricts ? Ce n’est pas pour éviter un biais de comparaisons multiples, qui ne produirait en théorie qu’une HSP aléatoire pour 1/(seuil Evalue) séquences requêtes. D’ailleurs les E-value ne sont généralement pas corrigées en fonction du nombre de requêtes. La raison pratique est que la similarité entre deux séquences est déjà bien faible à l’oeil pour une E-value de 10-5 : soit l’alignement correspondant est très court, soit il est plein de trous et a peu de résidus conservés. La raison théorique est que le modèle de protéines aléatoires employé pour calculer les scores BLAST n’est pas réaliste. Les protéines sont en réalité soumises à de nombreuses contraintes fonctionnelles, qui rendent impossibles la plupart des protéines aléatoires du modèle. Les acides aminés successifs ne sont pas indépendants car leurs propriétés physico-chimiques sont corrélées. Les protéines ont divers niveaux de structures, qui sont sélectionnés au cours des générations. L’espace que peuvent explorer les protéines réelles est donc beaucoup plus restreint que celui des protéines aléatoires du modèle de BLAST. Une paire de protéines réelles sans origine ancestrale commune peut donc potentiellement produire des HSP de faible E-value.
Il n’existe pas de modèle satisfaisant des protéines réelles, et donc pas d’arguments autres qu’empiriques pour choisir un seuil de E-value. Si l’on effectue une analyse à petite échelle, il est possible de privilégier la sensibilité de la recherche pour obtenir un maximum de résultats, quitte à éliminer les résultats douteux lors d’un examen individuel. En revanche lors d’analyses automatisées sur de grands jeux de données, il est gênant de ne pas avoir de critère de référence. Le seuil est souvent déterminé arbitrairement, par expérience ou pour les besoins de l’analyse subséquente, par exemple des HSP bien conservés pour réaliser des alignements multiples, ou des HSP en nombre raisonnable pour réaliser les calculs coûteux qui suivent.
Détecter et masquer les régions sensibles à la convergence évolutive
Si l’on cherche à identifier uniquement les régions homologues entre séquences avec BLAST, comment se prémunir des scores élevés produite par évolution convergente ? Avant toute chose, il est possible de filtrer les HSP identifiées selon plusieurs critères : E-value, pourcentage d’identité, proportion de sites identiques, longueur des alignements locaux.
Cela prémunit contre le fait que, globalement, les protéines sont plus similaires entre elles que le suppose le modèle de BLAST, mais ne permet pas d’éviter les cas de forte évolution convergente présentés ci-dessus. La stratégie générique pour éviter ces cas consiste à identifier les régions concernées dans les séquences, puis à modifier la façon dont elles sont considérées par l’algorithme d’alignement.
Une majorité des cas de forte évolution convergente est le fait de régions enrichies en certaines lettres ou contenant des répétitions simples [Frith et al., 2010], par exemple AAACAAAAAGAA, ATATATATAT, PPCDPPPKPPP. Ces régions sont dites de faible complexité, car elles sont très redondantes et contiennent peu d’information au sens de l’entropie de Shannon [Shannon, 1948]. Différents programmes ont été développés pour identifier ces régions, qui fonctionnent sur les séquences nucléiques (DUST), protéiques (SEG), ou les deux : TRF [Benson, 1999], TANTAN [Frith, 2011a]. Les régions non globulaires des protéines qui évoluent de façon convergente ne sont pas toujours bien détectées par les méthodes précédentes, qui n’identifient que les cas les plus évidents de complexité faible. Elles échouent notamment à éliminer de nombreuses régions transmembranaires, les hélices coiled-coil, les segments de peptides signal, qui sont très propices à des similarités nonhomologues.
Il faut employer des outils de détection spécifiques à chaque cas pour les identifier.
Alternatives à la comparaison de séquences avec BLASTP
Nous avons décrit une procédure générale pour comparer des séquences et éliminer les similarités qui ne proviendraient pas d’une origine ancestrale commune. Avant de s’interroger sur la façon dont nous allons exploiter ces relations entre séquences, il convient de préciser qu’il est parfois possible d’utiliser d’autres informations pour étudier leurs histoires communes. Les séquences que nous allons considérer sont des gènes, qui auront souvent été délimités dans des séquences plus longues (contig, génome). Ces gènes sont donc en relation physique avec d’autre gènes, de par leur succession le long d’un même support, dont ils partagent une part de l’histoire évolutive. Nous n’exploiterons pas directement cette information contextuelle, qui n’est d’ailleurs pas disponible si l’on étudie des données de transcriptomique ou de métagénomique.
Nous avons par ailleurs insisté sur le fait que nous comparions les textes des séquences génétiques. Il serait envisageable de comparer plutôt leur conformation tridimensionnelle, locale (structure secondaire) ou globale (structure tertiaire). La structure 3D d’une protéine est en effet bien mieux conservée que sa séquence primaire. Cependant il n’est pas possible d’acquérir directement de telles structures en grande quantité. Il est également difficile de prédire ces structures à partir des séquences (problème qui n’a d’ailleurs pas toujours une solution unique puisque les protéines peuvent changer de conformation 3D).
Il est en revanche courant d’étudier la structure en domaines des protéines. Cela consiste à identifier les domaines par des alignements locaux contre des domaines de référence, puis de comparer la composition en domaines des protéines. D’une part ce problème est d’une nature un peu différente, similaire à l’étude de la composition en gènes des génomes, d’autre part il suppose un modèle d’organisation des protéines en domaines, dont nous allons nous passer car il recoupe l’objet de notre étude (évolution combinatoire au sein des gènes).
Finalement, il nous reste pour les séquences codant pour des protéines l’alternative de comparer les versions en acides nucléiques, ou les versions traduites en acides aminés. Nous choisirons classiquement de comparer les séquences protéiques, car elles sont mieux conservées que les séquences nucléiques, ce qui est préférable à la grande échelle évolutive à laquelle nous travaillerons. Elles permettent de retrouver des relations plus anciennes entre séquences. Nous utiliserons donc essentiellement le programme BLASTP de la suite BLAST.
Des programmes tels que PSI-BLAST permettent de détecter des relations entre séquences plus anciennes encore que celles détectées par BLASTP. PSI-BLAST identifie pour cela une famille de gènes similaires au gène requête, détermine un modèle statistique de cette famille, et s’en sert pour identifier de nouvelles séquences correspondants au modèle.
Cette méthodologie repose donc sur la construction implicite de famille de gènes, ce qui recoupe l’objet de notre étude. Nous nous en tiendrons donc à la comparaison paire à paire des séquences par alignements locaux, afin de limiter les aprioris théoriques sur l’évolution des séquences.
Visualisation d’un réseau de similarité de séquences
Distances dans la projection du réseau
Après avoir comparé des séquences avec BLAST et transformé le résultat en un réseau de similarité de séquence, la première chose que l’on souhaite généralement faire est de le visualiser. Parmi les logiciels de visualisation de réseaux, les plus pratiques sont pour nous Cytoscape [Shannon et al., 2003] et Gephi [Bastian et al., 2009]. Ces deux logiciels acceptent en entrée un réseau décrit par une liste d’arêtes tabulée, format de notre sortie BLAST épurée.
Diverses méthodes d’agencement (layout) des noeuds et des arêtes sont proposées par ces logiciels. Il est recommandé d’en essayer plusieurs, et de faire varier leurs paramètres pour obtenir différentes projections d’un réseau. Les layouts Force-Directed, Edge-Weighted Spring Embedded et Organic sont les plus pratiques dans Cytoscape, tandis que le layout ForceAtlas2 est rapide et facilement paramétrable dans Gephi. Le fonctionnement précis de ces layouts diffère, mais ils sont généralement décrits par la même analogie mécanique, d’un réseau comportant des poids sur les noeuds et des ressorts sur les arêtes qu’il s’agit de faire converger vers un état d’énergie minimale.
Certains layouts peuvent prendre en compte une valeur numérique associée aux arêtes, qui module la force de rappel des ressorts. On utilise dans ce cas le Bit score comme valeur numérique ou la transformée logarithmique de la E-value (-log(E-value)), plutôt que la Evalue qui s’étale sur plusieurs centaines d’ordres de grandeurs. Si elle mérite d’être essayée et peut fournir des résultats intéressants, cette modulation du poids des arêtes n’est pas nécessaire, car la distance entre séquence se reflète déjà dans la topologie du réseau. Ce n’est pas une arête unique qui modifie fortement le placement des noeuds, mais la multiplicité des arêtes entre groupes de séquences similaires qui favorise l’émergence de structures dans la représentation finale.
La visualisation graphique d’un réseau met concrètement en pratique l’idée que l’on n’étudie pas des entités individuelles indépendantes, mais un ensemble structuré de relations. Ainsi la distance entre deux séquences dans une visualisation graphique ne représente a priori pas une distance évolutive portée par les arêtes, mais émerge de la prise en compte des similarités entre toutes les séquences. Un résultat frappant obtenu par Atkinson et ses collaborateurs [Atkinson et al., 2009] est que, même en exploitant uniquement la topologie du réseau, les distances 2D entre séquences dans une représentation graphique sont fortement corrélées aux distances en terme de E-value (coefficients de corrélation de 0.83 à 0.94 dans leurs analyses). Ils observent de plus que cette corrélation est peu sensible aux données manquantes, et qu’elle ne dépend pas de la taille du réseau mais davantage du seuil d’E -value employé. Ainsi la visualisation d’un réseau de similarité de séquences permet effectivement de voir les distances entre séquences étudiées1.
Effet des seuils de similarité sur la structure du réseau
La structure du réseau est fortement influencée par le niveau de similarité requis pour connecter les séquences. Le niveau de similarité de base est donné au moment de la comparaison BLAST, via le choix du seuil d’E-value maximal pour retourner un HSP. Il est possible de diminuer a posteriori ce seuil, et de filtrer les HSP sur d’autres valeurs, comme leur pourcentage d’identité ou leur longueur (exprimée en nombre de résidus ou en proportion des séquences couvertes). Ces filtres diminuent le nombre d’arêtes considérées (Figure 1-11) et la densité de connexions dans le réseau. De nouvelles structures locales apparaissent lorsqu’on ne conserve que les connexions entre séquences très similaires, jusqu’à éventuellement déconnecter certaines parties du réseau et créer de nouvelles composantes connexes.
Algorithmes de réseaux pour former des familles homologues
La construction de groupe de gènes homologues est une démarche fréquente, notamment dans l’optique de construire des alignements multiples puis des arbres phylogénétiques. Les différents algorithmes utilisés pour former des groupes de séquences homologues à partir d’une comparaison BLAST s’expriment naturellement à partir d’un réseau de similarité. Les réseaux ont parfois été utilisés pour concevoir ces algorithmes, mais sont souvent peu exploités pour les expliquer, et rarement utilisés pour visualiser leurs résultats. Nous proposons d’illustrer l’intérêt des réseaux de similarité de séquences sur ce problème classique. Nous présenterons ainsi les difficultés que rencontrent ces algorithmes, qui seront notre point de départ pour étudier les phénomènes combinatoires d’évolution des séquences.
Les réseaux permettent de construire des familles homologues divergentes
Une hypothèse centrale des méthodes de construction de familles homologues est qu’une similarité entre deux séquences implique leur homologie, c’est-à-dire qu’elles ont une origine ancestrale commune. Cette hypothèse est raisonnable dans la mesure où l’on emploie des seuils de similarité suffisamment élevés sur la E-value et le pourcentage d’identité, et que l’on masque les régions propices à la convergence évolutive lors des alignements. En revanche la réciproque de cette hypothèse est fausse : certaines séquenceshomologues ont trop divergé depuis leur séquence ancestrale commune et ne sont plus similaires. De telles séquences homologues mais non similaires sont appelées des homologues distants. Ainsi, deux séquences voisines dans un réseau de similarité de séquences peuvent être considérées homologues ; tandis que deux séquences qui ne sont pas voisines peuvent – ou non – être homologues.
Il est possible de retrouver le lien d’homologie entre des homologues distants, lorsqu’ils sont similaires aux mêmes séquences homologues intermédiaires. Cela correspond dans le réseau à considérer que les voisins (homologues) d’une séquence intermédiaire sont homologues entre eux (Figure 1-14.B). En reproduisant successivement ce principe, on peut retrouver les homologues distants d’une séquence via plusieurs homologues intermédiaires.
Un algorithme naturel pour former des familles homologues serait donc de considérer les composantes connexes du réseau de similarité de séquences. Deux séquences sont regroupées dans une même famille si et seulement si elles sont connectées dans le réseau directement ou par un chemin de similarité. Construire de telles familles homologues divergentes permet d’étudier l’évolution biologique à une plus grande échelle de temps.
1ère solution : Ne garder que les alignements locaux couvrant l’essentiel des séquences
Une réponse naturelle au problème de l’homologie partielle serait de ne s’intéresser qu’aux alignements locaux couvrant entièrement les séquences : deux séquences qui sont similaires – et s’alignent – sur toute leur longueur sont entièrement homologues. Un tel critère éliminerait les relations d’homologie partielles, mais aussi de nombreuses relations entre homologues complets. Il ne permettrait que de construire des familles homologues très conservées. Le rythme d’évolution est en effet souvent hétérogène le long des séquences : certaines régions sont conservées sur un grand nombre de générations, tandis que d’autres divergent rapidement.
Des critères moins stricts sont souvent employés, par exemple sur une proportion minimale des séquences couvertes par les alignements de 60 ou 80%. Si ces critères permettent d’étudier des relations entre homologues légèrement divergents, ils éliminent toujours les relations plus distantes et le problème de l’homologie partielle commence à se poser. Des tests empiriques ont montré que ce critère n’est pas un moyen fiable d’améliorer la reconstruction de familles homologues [Song et al., 2008].
2ème solution : étudier les protéines multi-domaines
Le phénomène d’homologie partielle remet en cause la conception des protéines comme des unités évolutives indivisibles. Une conception alternative est de considérer une protéine comme un assemblage de modules évolutifs indépendants, appelés domaines protéiques. Un domaine protéique est ici défini comme une sous-unité évolutive stable, qui peut être combiné avec d’autres modules pour former une grande diversité de protéines dîtes multi-domaines. Plusieurs approches utilisent ce modèle de protéine pour éviter le problème d’homologie partielle lors de la construction de familles homologues. Elles diffèrent dans leur méthodologie et dans leur conception de l’impact du phénomène d’homologie partielle.
La piste de GeneRAGE : étudier les familles mono-domaines
Une première approche consiste à s’intéresser aux protéines constituées d’un unique domaine évolutif, en considérant que les protéines multi-domaines sont des assemblages de protéines mono-domaines (Figure 1-15.A). Selon ce modèle, une protéine multi-domaine est similaire à plusieurs familles mono-domaines, et les connecte dans le réseau (Figure 1-15 29 .B).
L’algorithme GeneRAGE [Enright, Ouzounis, 2000] propose de détecter les protéines multidomaines1, de les retirer du réseau, pour pouvoir ensuite reconstruire les familles monodomaines sans les agglomérer.
|
Table des matières
Chapitre 1 – Introduction
1.1. L’abondance des données moléculaires ouvre de nouvelles questions évolutives
1.2. La science des réseaux pour l’étude des données relationnelles
1.3. La comparaison de séquences
1.4. Similarité, homologie et convergence évolutive
1.5. Réseaux de similarité de séquences
1.6. Objectifs de cette thèse
Chapitre 2 – Identification de familles de gènes composites
2.1. Processus combinatoires d’évolution des gènes
2.2. Identification des processus combinatoires dans les réseaux de similarité de séquences
Chapitre 3 – Problème de l’homologie : extension du champ des ressemblances informatives pour les évolutionnistes
3.1. L’homologie en biologie
3.2. Famille de gènes homologues
Chapitre 4 – Application de la notion d’air de famille à des jeux de données de virus
4.1. La diversité des virus est immense et très peu connue
4.2. Différentes classifications des virus
4.3. Evolution des virus
4.4. Etude systématique des phénomènes combinatoires chez les virus
4.5. Enjeux nouveaux abordés lors de cette étude
Chapitre 5 – Conclusion
5.1. Analyse des réseaux de similarité de séquences
5.2. Étude de l’évolution combinatoire des gènes
5.3. Pour une démarche pluraliste en évolution
Chapitre 6 – Bibliographie
Télécharger le rapport complet