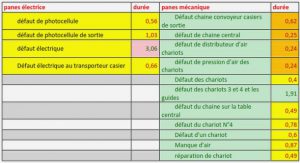
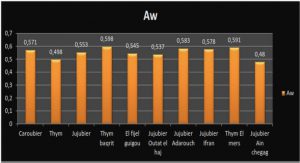
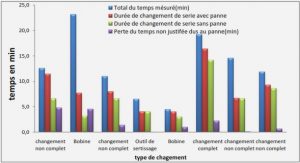
Premier théorème de Hohenberg et Kohn
Pour tout système, le potentiel externe ????, et donc l’énergie totale ?, est une fonction unique de la densité électronique n(?), c’est-à-dire E = E[n(?)] [22,26]. L’état fondamental d’un système composé d’un gaz d’électrons et sa densité de charge correspondante sont déterminés par le potentiel externe agissant sur ces électrons. Hohenberg et Kohn [11] ont déjà démontré que toutes les grandeurs physiques relatives à ce système (à l’instar de l’énergie totale du système) sont des fonctionnelles du potentiel externe, en considérant la correspondance biunivoque existant entre la densité électronique de l’état fondamental ?(?) et le potentiel externe ???? . En d’autres termes, en connaissant la densité de l’état fondamental, nous pouvons déduire le potentiel externe, et une fois le potentiel externe connu, nous pouvons résoudre l’équation de Schrödinger à plusieurs électrons et en fin tout savoir sur le système. Le but général de la théorie de la fonctionnelle de la densité est d’exprimer l’énergie du système quantique comme une fonctionnelle de la densité électronique au lieu de la fonction d’onde multiélectronique. C’est-à-dire, la base de la théorie de DFT est que toutes les observables d’un système multiélectronique sont fonctionnelles uniques de la densité électronique. A titre d’exemple, le potentiel externe à un corps ???? est le potentiel ???. Ceci signifie que le potentiel total à un corps de ce système représente l’interaction d’un électron avec le réseau cristallin
Implémentations pratiques de la DFT
Dans le but d’appliquer la théorie de la fonctionnelle de densité, selon l’approximation de BornOppenheimer, il était nécessaire d’utiliser une forme approximative de la fonctionnelle d’échange-corrélation. L’approximation fondamentale est celle du terme d’échangecorrélation, alors que les approximations supplémentaires, introduites par le traitement numérique, sont nécessaires pour résoudre les équations de Kohn et Sham (2.27), elles sont néanmoins maîtrisées par l’utilisation d’un code de calcul ab-initio. Dans la suite, une vue générale des principaux choix d’implémentation de la théorie DFT sera donnée, nous allons introduire aussi les deux approximations numériques : l’échantillonnage de la zone de Brillouin et l’énergie de coupure et en fin, la méthode de pseudopotentiel sera traitée en détail.
Méthode des pseudopotentiels
L’idée fondamentale d’un pseudopotentiel est le remplacement d’un potentiel avec un autre. L’application primaire en structure électronique est de remplacer le potentiel fort de coulomb du noyau et des effets des électrons étroitement liés au noyau (les électrons du cœurs) par un potentiel ionique efficace agissant sur les électrons de valence. L’approche pseudopotentielle (PP) utilise une description quantique pour les interactions électroniques dans le cadre de la DFT. Elle consiste en un couplage d’ondes planes et de pseudopotentiel au moyen d’une technique de transformée de Fourrier [22,26]. La famille des méthodes fonctionnelles de densité de Kohn-Sham se diffèrent par deux approches : dans la première on traite les électrons de cœur et de valence, appelée approche de tous-électron (AE ; all-electron), alors que la deuxième est appelée l’approximation de pseudopotentiels qui élimine l’effet des électrons du cœur. Toutes les deux approches sont intimement liées. Les calculs d’AE (All-Electron) sont généralement, en informatiques cher et excepte les systèmes avec un petit nombre atomique, puisqu’un grand nombre de fonctions de base sont exigées pour capturer le comportement oscillant de l’onde électronique fonctionnant près des noyaux. Lorsque le nombre d’électrons à traiter augmente, la résolution de l’équation de Schrödinger devient difficile à résoudre et les temps de calculs augmentent très vite. Par ailleurs, les propriétés physico-chimiques les plus étudiées dans les systèmes moléculaires ne font intervenir que les électrons de valence [22,26]. Dans un atome, seuls les électrons de périphériques (en petit nombre) contribuent à la formation des liaisons chimiques, tandis que les électrons du cœur (en grand nombre) sont eux fortement lies au noyau atomique et donc peu sensible à l’environnement proche de l’atome. Cette approximation, dite ‘frozen core approximation’, provient du fait que les électrons proches du noyau restent relativement inertes lorsque l’on change l’environnement chimique de l’atome (Ils ne changent pas d’état). L’avantage de cette approximation est que le nombre d’électron apparaissant d’une manière explicite dans les calculs soit réduit, donc le nombre d’équation à résoudre s’en trouve fortement réduit c’est à dire seuls les électrons de valence sont pris en compte [22,26]. Les fonctions d’onde relativement oscillantes dans la région de cœur (Fig. II.2), résultant de la contrainte d’orthogonalité avec les états de cœur (principe de répulsion de Pauli) et qui seraient relativement difficiles à décrire à partir d’une base d’ondes planes (nombre de vecteurs G très élevé), sont remplacées par des pseudo-fonctions d’onde qui sont dépourvues de nœuds dans la région du cœur (Fig. II.2). Le pseudopotentiel est de ce fait construit de manière à ce que les caractéristiques de déphasage qu’il produit sur les pseudo-fonctions d’onde soient identiques à celles résultant de l’action du cœur ionique sur les vraies fonctions d’onde de valence, tout en générant des pseudo-fonctions d’onde dépourvues d’oscillations (et par conséquent de nœuds) dans la région du cœur ionique. Au-delà de cette région du cœur ionique, délimitée par ?? (dite Rayon de coupure) (Fig. II.2), les pseudo-fonctions d’onde doivent être identiques aux fonctions d’onde de valence vraies, qui sont également appelées, par référence à ce modèle de pseudopotentiel, fonctions d’onde de valence « tous électrons » [22,26].
Conclusion générale
Les propriétés structurales et élastiques des pérovskites cubiques LaAlO3, PbTiO3, SrTiO3, BaTiO3, LaTiO3 et LaCrO3 ont été calculées par la théorie de la fonctionnelle de la densité (Density Functional Theory ou DFT) et les pseudopotentiels, en traitant l’énergie d’échange et de corrélation par l’approximation du gradient généralisé (Generalized Gradient Approximation ou GGA). Nous avons également utilisé l’approximation de Green et du potentiel écranté (Green and Screened Potential ou simplement GW) pour étudier les propriétés électroniques de ces pérovskites. Notre manuscrit contient une revue aussi complète que possible des travaux théoriques sur la théorie des perturbations à N-corps et l’approximation GW. En effet, nous avons développé des algorithmes et des programmes en C/C++ pour réduire le nombre des diagrammes de Feynman qui sont très utilisés pour simplifier et calculer la Self-énergie pendant l’exécution de la procédure de l’approximation GW. Ces diagrammes peuvent être illustrés par notre code créé par le langage Mathematica. Nous avons à cet effet proposé un algorithme pour calculer les intégrales curvilignes en utilisant les arbres couvrant à la place du calcul traditionnel de ces intégrales. Cette méthode réduit le temps d’exécution de l’approximation GW de ?! vers 4?−1. Les propriétés structurales et élastiques des pérovskites ont été étudiées dans ce travail, tels que le paramètre de maille à l’équilibre (?), le module de compression à l’équilibre (?), et le facteur d’anisotropie élastique (?). Le calcul de ces paramètres est en bon accord avec ceux qui sont obtenus expérimentalement. La méthode utilisée dans cette étude est très importante et efficace puisqu’elle permette de trouver les paramètres des propriétés structurales et élastiques des pérovskites dans le cas où aucun résultat expérimental de ces propriétés (exemple : LaTiO3 et LaCrO3). Notons que le facteur d’anisotropie élastique (?) obtenu augmente progressivement dans les composés : SrTiO3, PbTiO3, LaAlO3, BaTiO3, LaCrO3, LaTiO3 respectivement. En ce qui concerne l’étude des propriétés électroniques, nous avons calculé la bande d’énergie de ces pérovskites par l’approximation GW. Un désaccord GW avec l’approximation GGA et LDA apparait dans nos résultats. Lesquels résultats sont en bon accord avec ceux de l’expérience qui varient entre 1.01 et 1.23, et aussi avec le calcul théorique hybride B3LYB, HSE et GW+U. Il est à noter qu’il n’y a pas suffisamment de résultats expérimentaux concernant le calcul de l’énergie de gap pour pouvoir faire une bonne comparaison avec nos résultats. Les effets relativistes sur les orbitales atomiques et leurs conséquences sur la structure électronique sont négligés dans cette étude et ceci influe sur nos résultats. Nous trouvons au final que notre algorithme de calcul des intégrales curvilignes dans l’approximation GW réduit le temps d’exécution et que les valeurs obtenues de l’énergie de gap sont en bon accord avec les résultats expérimentaux disponibles. La méthode de GGA avec pseudopotentiel est donc performante pour l’étude des propriétés structurales et élastiques alors que l’approximation GW l’est pour l’étude des propriétés électroniques. Notons que l’approximation GW nécessite un temps d’exécution très long. Pour diminuer ce temps, nous proposons un algorithme basé sur les arbres couvrants à la place des intégrales curvilignes. Ces dernières représentent une partie importante de l’approximation GW.
|
Table des matières
Introduction générale
Chapitre I. Généralités sur la structure des pérovskites
I.1. Introduction
I.2. Description de la structure
I.3. Facteur de tolérance de Goldschmidt
I.4. Applications
I.5. Références
Chapitre II. Théorie de la fonctionnelle de la densité (DFT)
II.1. Introduction
II.2. Equation de Schrödinger et problème à plusieurs corps
II.2.1. Approximation de Born-Oppenheimer
II.2.2. Densité électronique
II.2.3. Modèle de Thomas-Fermi
II.3. Premier théorème de Hohenberg et Kohn
II.4. Deuxième théorème de Hohenberg et Kohn
II.5. Equations de Kohn-Sham
II.6. Fonctionnelles d’échange-corrélation
II.6.1. Approximation de la densité locale (LDA)
II.6.2. Approximation du gradient généralisé (GGA)
II.6.3. Au-delà de l’approximation du gradient généralisé, l’échelle de Jacob
II.7. Implémentations pratiques de la DFT
II.7.1. Panorama des principaux choix d’implémentation
II.7.2. Théorème de Bloch et bases d’ondes planes
II.7.3. Méthode des pseudopotentiels
II.8. Référence
Chapitre III. Théorie des perturbations à N-corps (MBPT)
III.1. Introduction
III.2. Seconde quantification
III.3. Théorie de perturbation
III.3.1. Théorème de Wick
III.3.2. Représentation de Hugenholtz
III.3.3. Méthode de Gaudin
III.3.4. Diagrammes connectés et déconnectés
III.3.5. Potentiel grand canonique
III.3.6. Diagrammes essentiellement distincts
III.4. Algorithme pour les diagrammes de Hugenholtz
III.4.1. Cycles
III.4.2. Etapes de l’algorithme
III.4.3. Description du programme
III.4.4. Résultats
III.5. Diagrammes réductibles et irréductibles
III.6. Equation de Dyson
III.7. Représentation de Lehmann
III.8. Equations de Hedin
III.9. L’approximation GW
III.10. Méthode de calcul du gap dans l’approximation GW
III.10.1. Génération de la structure électronique Kohn Sham
III.10.2. Génération de la matrice diélectrique
III.10.3. Calcul de Gap
III.11. Méthode d’intégration
III.12. Algorithme pour évaluer les diagrammes
III.13. Détails techniques
III.14. Références
Chapitre VI. Résultats et discussions
VI.1. Introduction
VI.2. Méthode de calcul
VI.3. Propriétés structurales et élastiques
VI.4. Propriétés électroniques
VI.4.1. Méthode et paramètres de calcul
VI.5. Références
Conclusion générale
Annexes
Télécharger le rapport complet